16.1. Comparative effectiveness research
16.1.1. Introduction
Comparative effectiveness research (CER) is designed to inform healthcare decisions for the prevention, the diagnosis and the treatment of a given health condition. CER therefore compares the potential benefits and harms of therapeutic strategies available in routine practice. The compared interventions may be related to similar treatments, such as competing medicines within the same class or with different mechanism of actions, or to different therapeutic approaches, such as surgical procedures and drug therapies. The comparison may focus only on the relative medical benefits and risks of the different options, or it may weigh both their costs and their benefits. The methods of comparative effectiveness research (Annu Rev Public Health 2012;33:425-45) defines the key elements of CER as a) a head-to-head comparison of active treatments, b) study population typical of the day-to-day clinical practice, and c) evidence focussed on informing healthcare and tailored to the characteristics of individual patients. CER is often discussed in the regulatory context of real-world evidence (RWE) generated by clinical trials or non-interventional (observational) studies using real-world data (RWD) (see Chapter 16.6).
The term ‘Relative effectiveness assessment (REA)’ is also used when comparing multiple technologies or a new technology against standard of care, while ‘rapid’ REA refers to performing an assessment within a limited timeframe in the case of a new marketing authorisation or a new indication granted for an approved medicine (see What is a rapid review? A methodological exploration of rapid reviews in Health Technology Assessments, Int J Evid Based Healthc. 2012;10(4):397-410).
16.1.2. Methods for comparative effectiveness research
CER may use a variety of data sources and methods. Methods to generate evidence for CER are divided below in four categories according to the data source: randomised clinical trials (RCTs), observational data, synthesis of published RCTs and cross-design synthesis.
16.1.2.1. CER based on randomised clinical trials
RCTs are considered the gold standard for demonstrating the efficacy of medicinal products but they rarely measure the benefits, risks or comparative effectiveness of an intervention in post-authorisation clinical practice. Moreover, relatively few RCTs are designed with an alternative therapeutic strategy as a comparator, which limits the utility of the resulting data in establishing recommendations for treatment choices. For these reasons, other methodologies such as pragmatic trials and large simple trials may be used to complement traditional confirmatory RCTs in CER. These trials are discussed in Chapter 4.2.7. The estimand framework described in the ICH E9-R1 Addendum on Estimands and Sensitivity Analysis in Clinical Trials to the Guideline on Statistical Principles for Clinical Trials (2019) should be considered in the planning of comparative effectiveness trials as it provides coherence and transparency on important elements of CER, namely definitions of exposures, endpoints, intercurrent events (ICEs), strategies to manage ICEs, approach to missing data and sensitivity analyses.
In order to facilitate comparison of results of CER between clinical trials, the COMET (Core Outcome Measures in Effectiveness Trials) Initiative aims at developing agreed minimum standardized sets of outcomes (‘core outcome sets’, COS) to be assessed and reported in effectiveness trials of a specific condition. Choosing Important Health Outcomes for Comparative Effectiveness Research: An Updated Review and User Survey (PLoS One 2016;11(1):e0146444) provides an updated review of studies that have addressed the development of COS for measurement and reporting in clinical trials. It is also worth noting that regulatory disease guidelines also establish outcomes of clinical interest to assess if a new therapeutic intervention works. Use of the same endpoint across RCTs thus facilitate comparisons.
16.1.2.2. CER using observational data
Use of observational data in CER
Although observational data from Phase IV trials, post-authorisation safety studies (PASS), or other RWD sources can be used to assess comparative effectiveness (and safety), it is generally inappropriate to use such data as a replacement for randomised evidence, especially in a confirmatory setting. Emulation of Randomized Clinical Trials With Nonrandomized Database Analyses: Results of 32 Clinical Trials (JAMA 2023;329(16):1376-85) concludes that RWE studies can reach similar conclusions as RCTs when design and measurements can be closely emulated, but this may be difficult to achieve. Concordance in results varied depending on the agreement metric. Emulation differences, chance, and residual confounding can contribute to divergence in results and are difficult to disentangle. When and How Can Real World Data Analyses Substitute for Randomized Controlled Trials? (Clin Pharmacol. Ther. 2017;102(6):924-33) suggests that RWE may be preferred over randomised evidence when studying a highly promising treatment for a disease with no other available treatment and where ethical considerations may preclude randomising patients to placebo, particularly if the disease is likely to result in severely compromised quality of life or mortality. In these cases, RWE could support medicines regulation by providing evidence on the safety and effectiveness of the therapy against the typical disease progression observed in the absence of treatment. This comparator disease trajectory may be assessed from historical controls that were diagnosed prior to the availability of the new treatment, or other sources.
When Can We Rely on Real-World Evidence to Evaluate New Medical Treatments? (Clin Pharmacol Ther. 2021; 111(1): 30–4) recommends that decisions regarding use of RWE in the evaluation of new treatments should depend on the specific research question, characteristics of the potential study settings and characteristics of the settings where study results would be applied, and take into account three dimensions in which RWE studies might differ from traditional clinical trials: use of RWD, delivery of real-world treatment and real-world treatment assignment. Observational data have, for instance, been used in proof-of-concept studies on anaplastic lymphoma kinase-positive non-small cell lung cancer, in pivotal trials on acute lymphoblastic leukaemia, thalassemia syndrome and haemophilia A, and in studies aimed at label expansion for epilepsy (see Characteristics of non-randomised studies using comparisons with external controls submitted for regulatory approval in the USA and Europe: a systematic review, BMJ Open. 2019;1;9(2):e024895; The Use of External Controls in FDA Regulatory Decision Making, Ther Innov Regul Sci. 2021;55(5):1019–35; and Application of Real-World Data to External Control Groups in Oncology Clinical Trial Drug Development, Front Oncol. 2022;11:695936).
Outside of specific circumstances, observational data and clinical trials are considered complementary to generate comprehensive evidence. For example, clinical trials may include historical controls from observational studies, or identify eligible study participants from disease registries. In defense of pharmacoepidemiology--embracing the yin and yang of drug research (N Engl J Med 2007;357(22):2219-21) shows that strengths and weaknesses of RCTs and observational studies may make both designs necessary in the study of drug effects. Hybrid approaches for CER allow to enrich clinical trials with observational data, for example:
-
Use of historical data to partially replace concurrent controls in randomised trials (see A roadmap to using historical controls in clinical trials - by Drug Information Association Adaptive Design Scientific Working Group (DIA-ADSWG), Orphanet J Rare Dis. 2020;15:69);
-
Use of historical data as prior evidence for relative treatment effects (see Prior Elicitation for Use in Clinical Trial Design and Analysis: A Literature Review, Int J Environ Res Public Health 2021;18(4):1833);
-
Construction of external control groups in single arm studies and Phase IV trials (see the draft FDA guidance Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products (2023), A Review of Causal Inference for External Comparator Arm Studies (Drug Saf. 2022;45(8):815-37) and Methods for external control groups for single arm trials or long-term uncontrolled extensions to randomized clinical trials, Pharmacoepidemiol Drug Saf. 2020; 29(11):1382–92).
Methods for CER using observational data
The use of non-randomised data for causal inference is notoriously prone to various sources of bias. For this reason, it is strongly recommended to carefully design or select the source of RWD and to adopt statistical methods that acknowledge and adjust for major risks of bias (e.g. confounding, missing data).
A framework to address these challenges adopts counterfactual theory to treat the observational study as an emulation of a randomised trial. Target trial emulation (described in Chapter 4.2.6.) is a strategy that uses existing tools and methods to formalise the design and analysis of observational studies. It stimulates investigators to identify potential sources of concerns and develop a design that best addresses these concerns and the risk of bias.
Target trial emulation consists in designing first a hypothetical ideal randomised trial (“target trial”) that would answer the research question. A second step identifies how to best emulate the design elements of the target trial (including its eligibility criteria, treatment strategies, assignment procedure, follow-up, outcome, causal contrasts and pre-specified analysis plan) using the available observational data source and the analytic approaches to apply, given the trade-offs in an observational setting. This approach may prevent some common biases, such as immortal time bias or prevalent user bias while also identifying situations where adequate emulation may not be possible using the data at hand. Emulating a Target Trial of Interventions Initiated During Pregnancy with Healthcare Databases: The Example of COVID-19 Vaccination (Epidemiology 2023;34(2):238-46) describes a step-by-step specification of the protocol components of a target trial and their emulation including sensitivity analyses using negative controls to evaluate the presence of confounding and, alternatively to a cohort design, a case-crossover or case-time-control design to eliminate confounding by unmeasured time-fixed factors. Comparative Effectiveness of BNT162b2 and mRNA-1273 Vaccines in U.S. Veterans (N Engl J Med. 2022;386(2):105-15) used target trial emulation to design a study where recipients of each vaccine were matched in a 1:1 ratio according to their baseline risk factors. This design could not be applied where baseline measurements are not collected at treatment start, which may be the case in some patient registries. Use of the estimand framework of the ICH E9 (R1) Addendum to design the target trial may increase transparency on the choices and assumptions needed in the observational study to emulate key trial protocol components, such as the estimand, exposure, intercurrent events (and the strategies to manage them), the missing data and the sensitivity analyses, and therefore may help evaluate the extent to which the observational study addresses the same question as the target trial. Studies on the effect of treatment duration are also often impaired by selection bias: How to estimate the effect of treatment duration on survival outcomes using observational data (BMJ. 2018;360: k182) proposes a 3-step approach (cloning, censoring, weighting) that could be used with target trial simulation to achieve better comparability with the treatment assignment performed in the trial and overcome bias in the observational study.
Statistical inference methods that can be used for conducting causal inference in non-interventional studies are described in Chapter 6.2.3 and include multivariable regression (to adjust for confounding, missing data, measurement error, and other sources of bias), propensity score methods (to adjust for confounding bias), prognostic or disease risk score methods (to adjust for confounding), G-methods and marginal structure models (to adjust for time-dependent confounding), and imputation methods (to adjust for missing data). In some situations, these methods can also be used to adjust for instrumental variables or to estimate prior event rate ratios. Causal Inference in Oncology Comparative Effectiveness Research Using Observational Data: Are Instrumental Variables Underutilized? (J Clin Oncol. 2023;41(13):2319-2322) summarises the key assumption, advantages and disadvantages of methods of causal inference in CER to adjust for confounding, including regression adjustment, propensity scores, difference-in differences, regression discontinuity and instrumental variable, highlighting that different methods can be combined. In some cases, observational studies may substantially benefit from collecting instrumental variables, and this should be considered early on when designing the study. For example, Dealing with missing data using the Heckman selection model: methods primer for epidemiologists (Int J Epidemiol. 2023;52(1):5-13) illustrates the use of instrumental variables to address data that are missing not at random. Another example is discussed in Association of Osteoporosis Medication Use After Hip Fracture With Prevention of Subsequent Nonvertebral Fractures: An Instrumental Variable Analysis (JAMA Netw Open. 2018;1(3):e180826.), where instrumental variables are used to adjust for unobserved confounders.
The Agency for Healthcare Research and Quality (AHRQ)’s Developing a Protocol for Observational Comparative Effectiveness Research: A User’s Guide (2013) identifies minimal standards and best practices for observational CER. It provides principles on a wide range of topics for designing research and developing protocols, with relevant questions to be addressed and checklists of key elements to be considered. The RWE Navigator website discusses methods using observational RWD with a focus on effectiveness research, such as the source of RWD, study designs, approaches to summarising and synthesising the evidence, modelling of effectiveness and methods to adjust for bias and governance aspects. It also presents a glossary of terms and case studies.
A roadmap to using historical controls in clinical trials - by Drug Information Association Adaptive Design Scientific Working Group (DIA-ADSWG) (Orphanet J Rare Dis. 2020;15:69) describes methods to minimise disadvantages of using historical controls in clinical trials, i.e. frequentist methods (e.g. propensity score methods and meta-analytical approach) or Bayesian methods (e.g. power prior method, adaptive designs and the meta-analytic combined [MAC] and meta-analytic predictive [MAP] approaches for meta-analysis). It also provides recommendations on approaches to apply historical controls when they are needed while maximising scientific validity to the extent feasible.
In the context of hybrid studies, key methodological issues to be considered when combining RWD and RCT data include:
-
Differences between the RWD and RCT in terms of data quality and applicability,
-
Differences between available RWD sources (e.g., due to heterogeneity in studied populations, differences in study design, etc.),
-
Risk of bias (particularly for RWD),
-
Generalisability (especially for RCT findings beyond the overall treatment effect).
Methods for systematic reviews and meta-analyses of observational studies are presented in Chapter 10 and Annex 1 of this Guide. They are also addressed in the Cochrane Handbook for Systematic Reviews of Interventions and the Methods Guide for Effectiveness and Comparative Effectiveness Reviews presented in section 16.1.2.3 of this Chapter.
Assessment of observational studies used in CER
Given the potential for bias and confounding in CER based on observational non-randomised studies, the design and results of such studies need to be adequately assessed. The Good ReseArch for Comparative Effectiveness (GRACE) (IQVIA, 2016) provides guidance to enhance the quality of observational CER studies and support their evaluation for decision-making using the provided checklist. How well can we assess the validity of non-randomised studies of medications? A systematic review of assessment tools (BMJ Open 2021;11:e043961) examined whether assessment tools for non-randomised studies address critical elements that influence the validity of findings from non-randomised studies for CER. It concludes that major design-specific sources of bias (e.g., lack of new-user design, lack of active comparator design, time-related bias, depletion of susceptibles, reverse causation) and statistical assessment of internal and external validity are not sufficiently addressed in most of the tools evaluated, although these critical elements should be integrated to systematically investigate the validity of non-randomised studies on comparative safety and effectiveness of medications. The article also provides a glossary of terms, a description of the characteristics the tools and a description of methodological challenges they address.
Comparison of results of observational studies and RCTs
Even if observational studies are not appropriate to replace RCTs for many CER topics and cannot answer exactly the same research question, comparison of their results for a same objective is currently a domain of interest. The underlying assumption is that if observational studies consistently match the results of published trials and predict the results of ongoing trials, this may increase the confidence in the validity of future RWD analyses performed in the absence of randomised trial evidence. In a review of five interventions, Randomized, controlled trials, observational studies, and the hierarchy of research designs (N Engl J Med 2000;342(25):1887-92) found that the results of well-designed observational studies (with either a cohort or case-control design) did not systematically overestimate the magnitude of treatment effects. Interim results from the 10 first emulations reported in Emulating Randomized Clinical Trials With Nonrandomized Real-World Evidence Studies: First Results From the RCT DUPLICATE Initiative (Circulation 2021;143(10):1002-13) found that differences between the RCT and corresponding RWE study populations remained but the RWE emulations achieved a hazard ratio estimate that was within the 95% CI from the corresponding RCT in 8 of 10 studies. Selection of active comparator therapies with similar indications and use patterns enhanced the validity of RWE. Final results of this project are discussed in the presentation Lessons Learned from Trial Replication Analyses: Findings from the DUPLICATE Demonstration Project (Duke-Margolis Center for Health Policy Workshop, 10 May 2022). Emulation Differences vs. Biases When Calibrating Real-World Evidence Findings Against Randomized Controlled Trials (Clin Pharmacol Ther. 2020;107(4):735-7) provides guidance on how to investigate and interpret differences in treatment effect estimates from the two study types.
An important source of selection bias leading to discrepancies between results of observational studies and RCTs may be the use of prevalent drug users in the former. Evaluating medication effects outside of clinical trials: new-user designs (Am J Epidemiol 2003;158(9):915-20) explains the biases introduced by use of prevalent drug users and how a new-user (or incident user) design eliminate these biases by restricting analyses to persons under observation at the start of the current course of treatment. The incident user design in comparative effectiveness research (Pharmacoepidemiol Drug Saf. 2013; 22(1):1–6) reviews published CER case studies in which investigators had used the incident user design and discusses its strengths (reduced bias) and weaknesses (reduced precision of comparative effectiveness estimates). Unless otherwise justified, the incident user design should always be used.
16.1.2.3. CER based on evidence synthesis of published RCTs
The Cochrane Handbook for Systematic Reviews of Interventions (version 6.2, 2022) describes in detail the process of preparing and maintaining systematic reviews on the effects of healthcare interventions. Although its scope is focused on Cochrane reviews, it has a much wider applicability. It includes guidance on the standard methods applicable to every review (planning a review, searching and selecting studies, data collection, risk of bias assessment, statistical analysis, GRADE and interpreting results), as well as more specialised topics. The (GRADE) working group (Grading of Recommendations Assessment, Development, and Evaluation) offers a structured process for rating quality of evidence and grading strength of recommendations in systematic reviews, health technology assessment and clinical practice guidelines. The Methods Guide for Effectiveness and Comparative Effectiveness Reviews (AHRQ, 2018) provides resources supporting comparative effectiveness reviews. They are focused on the US Effective Health Care (EHC) programme and may therefore have limitations as regards their generalisability.
A pairwise meta-analysis of RCT results is used when the primary aim is to estimate the relative effect of two interventions. Network meta-analysis for indirect treatment comparisons (Statist Med. 2002;21:2313–24) introduces methods for assessing the relative effectiveness of two treatments when they have not been compared directly in a randomised trial but have each been compared to other treatments. Overview of evidence synthesis and network meta-analysis – RWE Navigator discussed methods and best practices and gives access to published articles on this topic. A prominent issue that has been overlooked by some systematic literature reviews and network meta-analyses is the fact that RCTs included in a network meta-analysis are usually not comparable with each other even though they all compared to placebo. Different screening and inclusion/exclusion criteria often create different patient groups, and these differences are rarely discussed in indirect comparisons. Before indirect comparison are performed, researchers should therefore check the similarity/differences between the RCTs.
16.1.2.4. CER based on cross-design synthesis
Decision-making should ideally be based on all available evidence, including both randomised and non-randomised studies, and on both individual patient data and published aggregated data. Clinical trials are highly suitable to investigate efficacy but less practical to study long-term outcomes or rare diseases. On the other hand, observational data offer important insights about treatment populations, long-term outcomes (e.g., safety), patient-reported outcomes, prescription patterns, active comparators, etc. Combining evidence from these two sources could therefore be helpful to reach certain effectiveness/safety conclusions earlier or to address more complex questions. Several methods have been proposed but are still experimental. The article Framework for the synthesis of non-randomised studies and randomised controlled trials: a guidance on conducting a systematic review and meta-analysis for healthcare decision making (BMJ Evid Based Med. 2022;27(2):109-19) uses a 7-step mixed methods approach to develop guidance on when and how to best combine evidence from non-randomised studies and RCTs to improve transparency and build confidence in summary effect estimates. It provides recommendations on the most appropriate statistical approaches based on analytical scenarios in healthcare decision making and highlights potential challenges for the implementation of this approach.
16.1.3. Methods for REA
Methodological Guidelines for Rapid REA of Pharmaceuticals (EUnetHTA, 2013) cover a broad spectrum of issues on REA. They address methodological challenges that are encountered by health technology assessors while performing rapid REA and provide and discuss practical recommendations on definitions to be used and how to extract, assess and present relevant information in assessment reports. Specific topics covered include the choice of comparators, strengths and limitations of various data sources and methods, internal and external validity of studies, the selection and assessment of endpoints and the evaluation of relative safety.
16.1.4. Specific aspects
16.1.4.1. Secondary use of data for CER
Electronic healthcare records, patient registries and other data sources are increasingly used in clinical effectiveness studies as they capture real clinical encounters and may document reasons for treatment decisions that are relevant for the general patient population. As they are primarily designed for clinical care and not research, information on relevant covariates and in particular on confounding factors may not be available or adequately measured. These aspects are presented in other chapters of this Guide (see Chapter 6, Methods to address bias and confounding; Chapter 8.2, Secondary use of data, and other chapters for secondary use of data in other contexts) but they need to be specifically considered in the context of CER. For example, the Drug Information Association Adaptive Design Scientific Working Group ( DIA-ADSWG) Roadmap to using historical controls in clinical trials (Orphanet J Rare Dis. 2020;15:69) describes the main sources of RWD to be used as historical controls, with an Appendix providing guidance on factors to be evaluated in the assessment of the relevance of RWD sources and resultant analyses.
16.1.4.2. Data quality
Data quality is essential to ensure the rigor of CER and secondary use of data requires special attention. Comparative Effectiveness Research Using Electronic Health Records Data: Ensure Data Quality (SAGE Research Methods, 2020) discusses challenges and share experiences encountered during the process of transforming electronic health record data into a research quality dataset for CER. This aspect and other quality issues are also discussed in Chapter 13 on Quality management.
In order to address missing information, some CER studies have attempted to integrate information from healthcare databases with information collected ad hoc from study subjects. Enhancing electronic health record measurement of depression severity and suicide ideation: a Distributed Ambulatory Research in Therapeutics Network (DARTNet) study (J Am Board Fam Med. 2012;25(5):582-93) shows the value of linking direct measurements and pharmacy claims data to data from electronic healthcare records. Assessing medication exposures and outcomes in the frail elderly: assessing research challenges in nursing home pharmacotherapy (Med Care 2010;48(6 Suppl):S23-31) describes how merging longitudinal electronic clinical and functional data from nursing home sources with Medicare and Medicaid claims data can support unique study designs in CER but pose many challenging design and analytic issues.
16.1.4.3. Transparency and reproducibility
Clear and transparent study protocols for observational CER should be used to support the evaluation, interpretation and reproducibility of results. Use of the HARPER protocol template (HARmonized Protocol Template to Enhance Reproducibility of hypothesis evaluating real-world evidence studies on treatment effects: A good practices report of a joint ISPE/ISPOR task force, Pharmacoepidemiol Drug Saf. 2023;32(1):44-55) is recommended to facilitate protocol development and addressing important design components. Public registration and posting of the protocol, disease and drug code lists, and statistical programming is strongly recommended to ensure that results from comparative effectiveness studies can be replicated using the same data and/or design, as emphasised in Journal of Comparative Effectiveness Research welcoming the submission of study design protocols to foster transparency and trust in real-world evidence (J Comp Eff Res. 2023;12(1):e220197). The HMA-EMA Catalogue of RWD studies and ClinicalTrials.gov should be used for this purpose.
16.2. Vaccine safety and effectiveness
16.2.1. Vaccine safety
16.2.1.1. General considerations
The book Vaccination Programmes | Epidemiology, Monitoring, Evaluation (Hahné, S., Bollaerts, K., & Farrington, P., Routledge, 2021) is a comprehensive textbook addressing most of the concepts presented in this Chapter. For contents related to safety monitoring of vaccines, it further builds on the 2014 ADVANCE Report on appraisal of vaccine safety methods that described a wide range of direct and indirect methods for vaccine safety assessment. Specific aspects related to vaccine safety and effectiveness are discussed in several documents:
-
The Report of the CIOMS/WHO Working Group on Definition and Application of Terms for Vaccine Pharmacovigilance (2012) provides definitions and explanatory notes for the terms ‘vaccine pharmacovigilance’, ‘vaccination failure’ and ‘adverse event following immunisation (AEFI)’.
-
The Guide to active vaccine safety surveillance: Report of CIOMS working group on vaccine safety – executive summary (Vaccine 2017;35(32):3917-21) describes the process for selecting the best approach to active surveillance considering key implementation issues, including in resource-limited countries.
-
The CIOMS Guide to Vaccine Safety Communication (2018) addresses vaccine safety communication aspects for regulators, vaccination policy-makers, and other stakeholders, when introducing vaccines in populations, based on selected examples.
-
The Brighton Collaboration provide a resource to facilitate and harmonise collection, analysis, and presentation of vaccine safety data, including case definitions for outcomes of interest, including adverse events of special interest (AESIs).
-
Module 4 (Surveillance) of the e-learning training course Vaccine Safety Basics of the World Health Organization (WHO) describes pharmacovigilance principles, causality assessment procedures, surveillance systems, and places safety in the context of the vaccine benefit/risk profile.
-
Recommendations on vaccine-specific aspects of the EU Pharmacovigilance System, including on risk management, signal detection and post-authorisation safety studies (PASS) are presented in Module P.I: Vaccines for prophylaxis against infectious diseases (EMA, 2013) of the Good Pharmacovigilance Practices (GVP).
-
The WHO Covid-19 vaccines: safety surveillance manual (WHO, 2020) was developed upon recommendation of the WHO Global Advisory Committee on Vaccine Safety (GACVS) and describes categories of surveillance strategies: passive, active, cohort event monitoring, and sentinel surveillance. While developed for COVID-19 vaccines, it can be used to guide pandemic preparedness activities for the monitoring of novel vaccines.
-
A vaccine study design selection framework for the postlicensure rapid immunization safety monitoring program (Am J Epidemiol. 2015;181(8):608-18) describes in a tabular form strengths and weaknesses of study designs and can be broadly applied to vaccine research questions beyond safety assessment.
16.2.1.2. Signal detection and validation
Besides a qualitative analysis of spontaneous case reports or case series, quantitative methods such as disproportionality analyses (described in Chapter 11) and observed-to-expected (O/E) analyses are routinely employed in signal detection and validation for vaccines. Several documents discuss the merits and review the methods of these approaches for vaccines.
Disproportionality analyses
GVP Module P.I: Vaccines for prophylaxis against infectious diseases describes aspects to be considered when applying methods for vaccine disproportionality analyses, including choice of the comparator group and use of stratification. Effects of stratification on data mining in the US Vaccine Adverse Event Reporting System (VAERS) (Drug Saf. 2008;31(8):667-74) demonstrates that stratification can reveal and reduce confounding and unmask some vaccine-event pairs not found by crude analyses. However, Stratification for Spontaneous Report Databases (Drug Saf. 2008;31(11):1049-52) highlights that extensive use of stratification in signal detection algorithms should be avoided, as it can mask true signals. Vaccine-Based Subgroup Analysis in VigiBase: Effect on Sensitivity in Paediatric Signal Detection (Drug Saf. 2012;35(4):335-46) further examines the effects of subgroup analyses based on the relative distribution of vaccine/non-vaccine reports in paediatric adverse drug reaction data (ADR) data. In Performance of Stratified and Subgrouped Disproportionality Analyses in Spontaneous Databases (Drug Saf. 2016;39(4):355-64), subgrouping by vaccines/non-vaccines resulted in a decrease in both precision and sensitivity in all spontaneous report databases that contributed data.
Optimization of a quantitative signal detection algorithm for spontaneous reports of adverse events post immunization (Pharmacoepidemiol Drug Saf. 2013;22(5): 477–87) explores various ways of improving performance of signal detection algorithms for vaccines.
Adverse events associated with pandemic influenza vaccines: comparison of the results of a follow-up study with those coming from spontaneous reporting (Vaccine 2011;29(3):519-22) reported a more complete pattern of reactions when using two complementary methods for first characterisation of the post-marketing safety profile of a new vaccine, which may impact on signal detection.
In Review of the initial post-marketing safety surveillance for the recombinant zoster vaccine (Vaccine 2020;38(18):3489-500), the time-to-onset distribution of zoster vaccine-adverse event pairs was used to generate a quantitative signal of unexpected temporal relationship.
Bayesian disproportionality methods have also been developed to generate disproportionality signals. In Association of Facial Paralysis With mRNA COVID-19 Vaccines: A Disproportionality Analysis Using the World Health Organization Pharmacovigilance Database (JAMA Intern Med. 2021;e212219), a potential safety signal for facial paralysis was explored using the Bayesian neural network method.
In Disproportionality analysis of anaphylactic reactions after vaccination with messenger RNA coronavirus disease 2019 vaccines in the United States (Ann Allergy Asthma Immunol. 2021; S1081-1206(21)00267-2) the CDC Wide-ranging Online Data for Epidemiologic Research (CDC WONDER) system was used in conjunction with proportional reporting ratios to evaluate whether rates of anaphylaxis cases reported in the VAERS database following administration of mRNA COVID-19 vaccines is disproportionately different from all other vaccines.
Signaling COVID-19 Vaccine Adverse Events (Drug Saf. 2022 Jun 23:1–16) discusses the extent, direction, impact, and causes of masking, an issue associated with signal detection methodologies, in which signals for a product of interest are hidden by the presence of other reported products, which may limit the understanding of the risks associated with COVID-19 and other vaccines, and delay their identification.
Observed-to-expected analyses and background incidence rates
In vaccine vigilance, O/E analyses compare the ‘observed’ number of cases of an adverse event occurring in vaccinated individuals and recorded in a data collection system (e.g. a spontaneous reporting system or an electronic healthcare database) and the ‘expected’ number of cases that would have naturally occurred in the same population without vaccination, estimated from available incidence rates in a non-vaccinated population. O/E analyses constitute a first step in the continuum of safety signal evaluation, and can guide further steps such as a formal pharmacoepidemiological study. GVP Module P.I: Vaccines for prophylaxis against infectious diseases (EMA, 2013) suggests conducting O/E analyses for signal validation and preliminary signal evaluation when prompt decision-making is required, and there is insufficient time to review a large number of individual cases. It discusses key requirements of O/E analyses: an observed number of cases detected in a passive or active surveillance system, near real-time exposure data, appropriately stratified background incidence rates calculated on a population similar to the vaccinated population (for the expected number of cases), the definition of appropriate risk periods (where there is suspicion and/or biological plausibility that there is a vaccine‐associated increased risk of the event) and sensitivity analyses around these measures. O/E analyses may require some adjustments for continuous monitoring due to inflation of type 1 error rates when multiple tests are performed. The method is further discussed in Pharmacoepidemiological considerations in observed‐to‐expected analyses for vaccines (Pharmacoepidemiol Drug Saf. 2016;25(2):215-22) and the review Near real‐time vaccine safety surveillance using electronic health records - a systematic review of the application of statistical methods (Pharmacoepidemiol Drug Saf. 2016;25(3):225-37).
O/E analyses require several pre-defined assumptions based on the requirements listed above. Each of these assumptions can be associated with uncertainties. How to manage these uncertainties is also addressed in Pharmacoepidemiological considerations in observed-to-expected analyses for vaccines (Pharmacoepidemiol Drug Saf. 2016;25(2):215–22). Observed-over-Expected analysis as additional method for pharmacovigilance signal detection in large-scaled spontaneous adverse event reporting (Pharmacoepidemiol Drug Saf. 2023;32(7):783-794) uses two examples of events of interest (idiopathic peripheral facial paralysis and Bell's palsy) in the context of the COVID-19 immunisation campaigns, when very large numbers of case safety reports (ICSRs) had to be timely handled.
Use of population based background rates of disease to assess vaccine safety in childhood and mass immunisation in Denmark: nationwide population based cohort study (BMJ. 2012;345:e5823) illustrates the importance of collecting background rates by estimating risks of coincident associations of emergency consultations, hospitalisations and outpatients consultations, with vaccination. Rates of selected disease events for several countries may vary by age, sex, method of ascertainment, and geography, as shown in Incidence Rates of Autoimmune Diseases in European Healthcare Databases: A Contribution of the ADVANCE Project (Drug Saf. 2021;44(3):383-95), where age-, gender-, and calendar-year stratified incidence rates of nine autoimmune diseases in seven European healthcare databases from four countries were generated to support O/E analyses. Guillain-Barré syndrome and influenza vaccines: A meta-analysis (Vaccine 2015; 33(31):3773-8) suggests that a trend observed between different geographical areas would be consistent with a different susceptibility of developing a particular adverse reaction among different populations. In addition, comparisons with background rates may be invalid if conditions are unmasked at vaccination visits (see Human papillomavirus vaccination of adult women and risk of autoimmune and neurological diseases, J Intern Med. 2018;283:154-65)).
Several studies have generated background incidence rates of AESIs for COVID-19 vaccines and discuss methodological challenges related to identifying AESIs in electronic health records (EHRs) (see The critical role of background rates of possible adverse events in the assessment of COVID-19 vaccine safety, Vaccine 2021;39(19):2712-18).
In Arterial events, venous thromboembolism, thrombocytopenia, and bleeding after vaccination with Oxford-AstraZeneca ChAdOx1-S in Denmark and Norway: population based cohort study (BMJ. 2021;373:n1114), observed age- and sex-specific rates of events among vaccinated people were compared with expected rates in the general population calculated from the same databases, thereby removing a source of variability between observed and expected rates. Where this is not possible, rates from multiple data sources have shown to be heterogeneous, and the choice of relevant data should take into account differences in database and population characteristics related to different diagnoses, recording and coding practices, source populations (e.g., inclusion of subjects from general practitioners and/or hospitals), healthcare systems, and linkage ability (e.g., to hospital records). This is further discussed in Characterising the background incidence rates of adverse events of special interest for covid-19 vaccines in eight countries: multinational network cohort study (BMJ. 2021;373:n1435) and Background rates of five thrombosis with thrombocytopenia syndromes of special interest for COVID-19 vaccine safety surveillance: Incidence between 2017 and 2019 and patient profiles from 38.6 million people in six European countries (Pharmacoepidemiol Drug Saf. 2022;31(5):495-510).
Contextualising adverse events of special interest to characterise the baseline incidence rates in 24 million patients with COVID-19 across 26 databases: a multinational retrospective cohort study (EClinicalMedicine. 2023;58:101932) used data from primary care, electronic health records, and insurance claims mapped to a common data model to characterise the incidence rates of AESIs, also following SARS-CoV-2 infection (considered a confounder), compared them to historical rates in the general population, and addressed issues of heterogeneity.
Historical comparator designs may generate false positives, as discussed in Bias, Precision and Timeliness of Historical (Background) Rate Comparison Methods for Vaccine Safety Monitoring: An Empirical Multi-Database Analysis (Front Pharmacol. 2021;12:773875), which explores the effect of empirical calibration on type 1 and 2 errors using outcomes presumed to be unrelated to vaccines (negative control outcomes) as well as positive controls (outcomes simulated to be caused by the vaccines).
Factors Influencing Background Incidence Rate Calculation: Systematic Empirical Evaluation Across an International Network of Observational Databases (Front Pharmacol. 2022;13:814198) examined the sensitivity of rates to the choice of design parameters using 12 data sources to systematically examine their influence on incidence rates using 15 AESIs for COVID-19 vaccines. Rates were highly influenced by the choice of the database (varying by up to a factor of 100), the choice of anchoring (e.g., health visit, vaccination, or arbitrary date) for the time-at-risk start, the choice of clean window and time-at-risk duration, but less so by secular or seasonal trends. It concluded that results should be interpreted in the context of study parameter choices.
Sequential methods
Sequential methods, as described in Early detection of adverse drug events within population-based health networks: application of sequential methods (Pharmacoepidemiol Drug Saf. 2007;16(12):1275-84), allow O/E analyses to be performed on a routine (e.g., weekly) basis using cumulative data with adjustment for multiplicity. Such methods are routinely used for near-real time surveillance in the Vaccine Safety Datalink (VSD) (see Near real-time surveillance for influenza vaccine safety: proof-of-concept in the Vaccine Safety Datalink Project, Am J Epidemiol 2010;171(2):177-88). Potential issues are described in Challenges in the design and analysis of sequentially monitored postmarket safety surveillance evaluations using electronic observational health care data (Pharmacoepidemiol Drug Saf. 2012;21(S1):62-71). A review of signals detected over 3 years in the VSD concluded that care with data quality, outcome definitions, comparator groups, and duration of surveillance, is required to enable detection of true safety issues while controlling for error (Active surveillance for adverse events: the experience of the Vaccine Safety Datalink Project, Pediatrics 2011;127(S1):S54-S64).
A new self-controlled case series method for analyzing spontaneous reports of adverse events after vaccination (Am J Epidemiol. 2013;178(9):1496-504) extends the self-controlled case series approach (see Chapter 4.2.3, and 16.2.2.2 in this Chapter) to explore and quantify vaccine safety signals from spontaneous reports using different assumptions (e.g., considering large amount of underreporting, and variation of reporting with time since vaccination). The method should be seen as a signal strengthening approach for quickly exploring a signal prior to a pharmacoepidemiological study (see for example, Kawasaki disease and 13-valent pneumococcal conjugate vaccination among young children: A self-controlled risk interval and cohort study with null results, PLoS Med. 2019;16(7):e100284).
The tree-based scan statistic (TreeScan) is a statistical data mining method that can be used for the detection of vaccine safety signals from large health insurance claims and electronic health records (Drug safety data mining with a tree-based scan statistic, Pharmacoepidemiol Drug Saf. 2013;22(5):517-23). A Broad Safety Assessment of the 9-Valent Human Papillomavirus Vaccine (Am J Epidemiol. 2021;kwab022) and A broad assessment of covid-19 vaccine safety using tree-based data-mining in the vaccine safety datalink (Vaccine. 2023;41(3):826-835) used the self-controlled tree-temporal scan statistic which does not require pre-specified outcomes or specific post-exposure risk periods. The method requires further evaluation of its utility for routine vaccine surveillance in terms of requirements for large databases and computer resources, as well as predictive value of the signals detected.
16.2.1.3. Study designs for vaccine safety assessment
A complete review of vaccine safety study designs and methods for hypothesis-testing studies is included in the ADVANCE Report on appraisal of vaccine safety methods (2014) and in Part IV of the book Vaccination Programmes | Epidemiology, Monitoring, Evaluation (Hahné, S., Bollaerts, K., & Farrington, P., Routledge, 2021).
Current Approaches to Vaccine Safety Using Observational Data: A Rationale for the EUMAEUS (Evaluating Use of Methods for Adverse Events Under Surveillance-for Vaccines) Study Design (Front Pharmacol. 2022;13:837632) provides an overview of strengths and limitations of study designs for vaccine safety monitoring and discusses the assumptions made to mitigate bias in such studies.
Methodological frontiers in vaccine safety: qualifying available evidence for rare events, use of distributed data networks to monitor vaccine safety issues, and monitoring the safety of pregnancy interventions (BMJ Glob Health. 2021;6(Suppl 2):e003540) addresses multiple aspects of pharmacoepidemiological vaccine safety studies, including study designs.
Cohort and case-control studies
There is a large body of published literature reporting on the use of the cohort design (and to a lesser extent, the case-control design) for the assessment of vaccine safety. Aspects of these designs presented in Chapters 4.2.1 and 4.2.2 are applicable to vaccine studies (for the cohort design, see also the examples of studies on background incidence rates in paragraph 16.2.2.1 of this Chapter). A recent illustration of the cohort design is provided in Clinical outcomes of myocarditis after SARS-CoV-2 mRNA vaccination in four Nordic countries: population based cohort study (BMJ Med. 2023 Feb 1;2(1):e000373) which used nationwide register data to compare clinical outcomes of myocarditis associated with vaccination, with COVID-19 disease, and with conventional myocarditis, with respect to readmission to hospital, heart failure, and death, using the Kaplan-Meier estimator approach.
Cohort-event monitoring
Prospective cohort-event monitoring (CEM) including active surveillance of vaccinated subjects using smartphone applications and/or web-based tools has been extensively used to monitor the safety of COVID-19 vaccines, as primary data collection was the only means to rapidly identify safety concerns when the vaccines started to be used at large scale. A definition of cohort-event monitoring is provided in The safety of medicines in public health programmes : pharmacovigilance, an essential tool (WHO, 2006, Chapter 6.5, Cohort event monitoring, pp 40-41). Specialist Cohort Event Monitoring studies: a new study method for risk management in pharmacovigilance (Drug Saf. 2015;38(2):153-63) discusses the rationale and features to address possible bias, and some applications of this design. COVID-19 vaccine waning and effectiveness and side-effects of boosters: a prospective community study from the ZOE COVID Study (Lancet Infect Dis. 2022:S1473-3099(22)00146-3) is a longitudinal, prospective, community-based study to assess self-reported systemic and localised adverse reactions of COVID-19 booster doses, in addition to effectiveness against infection (a confounder). Self-reported data may introduce information bias, as some participants might be more likely to report symptoms and some may drop out; however, multi-country CEM studies allow to include large populations, as shown in Cohort Event Monitoring of Adverse Reactions to COVID-19 Vaccines in Seven European Countries: Pooled Results on First Dose (Drug Saf. 2023;46(4):391-404).
Case-only designs
Traditional designs such as the cohort and case-control designs (see Chapters 4.2.1 and 4.2.2) may be difficult to implement in circumstances of high vaccine coverage (for example, in mass immunisation campaigns such as for COVID-19), a lack of an appropriate comparator group (e.g., unvaccinated), or a lack of adequate covariate information at the individual level. Frequent sources of confounding are underlying health status and factors influencing the likelihood of being vaccinated, such as access to healthcare or belonging to a high-risk group (see paragraph 16.2.4.1 on Studies in special populations in this Chapter). In such situations, case-only designs may provide stronger evidence than large cohort studies as they control for fixed individual-level confounders (such as demographics, genetics, or social deprivation) and have similar, sometimes higher, power (see Control without separate controls: evaluation of vaccine safety using case-only methods, Vaccine 2004;22(15-16):2064-70). Case-only designs are discussed in Chapter 4.2.3.
Several publications have compared traditional and case-only study designs for vaccine studies:
-
Epidemiological designs for vaccine safety assessment: methods and pitfalls (Biologicals 2012;40(5):389-92) used three designs (cohort, case-control, and self-controlled case-series (SCCS)) to illustrate aspects such as case definition, limitations of data sources, uncontrolled confounding, and interpretation of findings.
-
Comparison of epidemiologic methods for active surveillance of vaccine safety (Vaccine 2008; 26(26):3341-45) performed simulations to compare four designs (matched cohort, vaccinated-only (risk interval) cohort, case-control, and SCCS). The cohort design allowed for the most rapid signal detection, less false-positive error and highest statistical power in sequential analyses. However, one limitation of this simulation was the lack of case validation.
-
The simulation study Four different study designs to evaluate vaccine safety were equally validated with contrasting limitations (J Clin Epidemiol. 2006; 59(8):808-18) compared four designs (cohort, case-control, risk-interval and SCCS) and concluded that all were valid, however, with contrasting strengths and weaknesses. The SCCS, in particular, proved to be an efficient and valid alternative to the cohort design.
-
Hepatitis B vaccination and first central nervous system demyelinating events: Reanalysis of a case-control study using the self-controlled case series method (Vaccine 2007;25(31):5938-43) describes how the SCCS found similar results as the case-control design but with greater precision, based on the assumption that exposures are independent of earlier events, and recommended that case-series analyses should be conducted in parallel to case-control analyses.
It is increasingly considered good practice to use combined approaches, such as a cohort design and sensitivity analyses using a self-controlled method, as this provides an opportunity for minimising some biases that cannot be taken into account in the primary design (see for example, Myocarditis and pericarditis associated with SARS-CoV-2 vaccines: A population-based descriptive cohort and a nested self-controlled risk interval study using electronic health care data from four European countries; Front Pharmacol. 2022;13:1038043).
While the SCCS is suited to secondary use of data, it may not always be appropriate in situations where rapid evidence generation is needed, since follow-up time needs to be accrued. In such instances, design approaches include the SCRI method that can be used to shorten observation time (see The risk of Guillain-Barre Syndrome associated with influenza A (H1N1) 2009 monovalent vaccine and 2009-2010 seasonal influenza vaccines: Results from self-controlled analyses, Pharmacoepidemiol. Drug Saf 2012;21(5):546-52; and Chapter 4.2.3); O/E analyses using historical background rates (see Near real-time surveillance for influenza vaccine safety: proof-of-concept in the Vaccine Safety Datalink Project, Am J Epidemiol 2010;171(2):177-88); or traditional case-control studies (see Guillain-Barré syndrome and adjuvanted pandemic influenza A (H1N1) 2009 vaccine: multinational case-control study in Europe, BMJ 2011;343:d3908).
Nevertheless, the SCCS design is an adequate method to study vaccine safety, provided the main requirements of the method are taken into account (see Chapter 4.2.3). An illustrative example is shown in Bell's palsy and influenza(H1N1)pdm09 containing vaccines: A self-controlled case series (PLoS One. 2017;12(5):e0175539). In First dose ChAdOx1 and BNT162b2 COVID-19 vaccinations and cerebral venous sinus thrombosis: A pooled self-controlled case series study of 11.6 million individuals in England, Scotland, and Wales (PLoS Med. 2022;19(2):e1003927), pooled primary care, secondary care, mortality, and virological data were used. The authors discuss the possibility that the SCCS assumption of event-independent exposure may not have been satisfied in the case of cerebral venous sinus thrombosis (CVST) since vaccination prioritised risk groups, which may have caused a selection effect where individuals more likely to have an event were less likely to be vaccinated and thus less likely to be included in the analyses. In First-dose ChAdOx1 and BNT162b2 COVID-19 vaccines and thrombocytopenic, thromboembolic and hemorrhagic events in Scotland (Nat Med. 2021; 27(7):1290-7), potential residual confounding by indication in the primary analysis (a nested case-control design) was addressed by a SCCS to adjust for time-invariant confounders. Risk of acute myocardial infarction and ischaemic stroke following COVID-19 in Sweden: a self-controlled case series and matched cohort study (Lancet 2021;398(10300):599-607) showed that a COVID-19 diagnosis is an independent risk factor for the events, using two complementary designs in Swedish healthcare data: a SCCS to calculate incidence rate ratios in temporal risk periods following COVID-19 onset, and a matched cohort study to compare risks within 2 weeks following COVID-19 to the risk in the background population.
A modified self-controlled case series method for event-dependent exposures and high event-related mortality, with application to COVID-19 vaccine safety (Stat Med. 2022;41(10):1735-50) used data from a study of the risk of cardiovascular events, together with simulated data, to illustrate how to handle event-dependent exposures and high event-related mortality, and proposes a newly developed test to determine whether a vaccine has the same effect (or lack of effect) at different doses.
Estimating the attributable risk
The attributable risk of a given safety outcome (assuming a causal effect attributable to vaccination) is an important estimate to support public health decision-making in the context of vaccination campaigns. In the population-based cohort study Investigation of an association between onset of narcolepsy and vaccination with pandemic influenza vaccine, Ireland April 2009-December 2010 (Euro Surveill. 2014;19(17):15-25), the relative risk was calculated as the ratio of the incidence rates for vaccinated and unvaccinated subjects, while the absolute attributable risk was calculated as the difference in incidence rates. Safety of COVID-19 vaccination and acute neurological events: A self-controlled case series in England using the OpenSAFELY platform (Vaccine. 2022;40(32):4479-4487) used primary care, hospital admission, emergency care, mortality, vaccination, and infection surveillance data linked through a dedicated data analytics platform, and calculated the absolute risk of selected AESIs.
Case-coverage design
The case-coverage design is a type of ecological design using exposure information on cases, and population data on vaccination coverage to serve as control. It compares odds of exposure in cases to odds of exposure in the general population, similar to the screening method used in vaccine effectiveness studies (see below paragraph16.2.3.3 in this Chapter). However, it does not control for residual confounding and is prone to selection bias introduced by propensity to seek care (and vaccination) and by awareness of possible occurrence of a specific outcome, and does not consider underlying medical conditions, with limited comparability between cases and controls. In addition, it requires reliable and granular vaccine coverage data corresponding to the population from which cases are drawn, to allow control of confounding by stratified analyses (see for example, Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis, BMJ. 2013; 346:f794).
16.2.2. Vaccine effectiveness
16.2.2.1. General considerations
The book Vaccination Programmes | Epidemiology, Monitoring, Evaluation (Hahné, S., Bollaerts, K., & Farrington, P., Routledge, 2021) discusses the concept of vaccine effectiveness and provides further insight into the methods discussed in this section. The book Design and Analysis of Vaccine Studies (ME Halloran, IM Longini Jr., CJ Struchiner, Ed., Springer, 2010) presents methods and a conceptual framework of the different effects of vaccination at the individual and population level, and includes methods for evaluating indirect, total and overall effects of vaccination in populations.
A key reference is Vaccine effects and impact of vaccination programmes in post-licensure studies (Vaccine 2013;31(48):5634-42), which reviews methods for the evaluation of the effectiveness of vaccines and vaccination programmes and discusses design assumptions and biases to consider. A framework for research on vaccine effectiveness (Vaccine 2018;36(48):7286-93) proposes standardised definitions, considers models of vaccine failure, and provides methodological considerations for different designs.
Evaluation of influenza vaccine effectiveness: a guide to the design and interpretation of observational studies (WHO, 2017) provides an overview of methods to study influenza vaccine effectiveness, also relevant for other vaccines. Evaluation of COVID-19 vaccine effectiveness (WHO, 2021) provides guidance on how to monitor COVID-19 vaccine effectiveness using observational study designs, including considerations relevant to low- and middle-income countries. Methods for measuring vaccine effectiveness and a discussion of strengths and limitations are presented in Exploring the Feasibility of Conducting Vaccine Effectiveness Studies in Sentinel’s PRISM Program (CBER, 2018). Although focusing on the planning, evaluation, and modelling of vaccine efficacy trials, Challenges of evaluating and modelling vaccination in emerging infectious diseases (Epidemics 2021:100506) includes a useful summary of references for the estimation of indirect, total, and overall effects of vaccines.
16.2.2.2. Sources of exposure and outcome data
Data sources for vaccine studies largely rely on vaccine-preventable infectious disease surveillance (for effectiveness studies) and vaccine registries or vaccination data recorded in healthcare databases (for both safety and effectiveness studies). Considerations on validation of exposure and outcome data are provided in Chapter 5.
Infectious disease surveillance is a population-based, routine public health activity involving systematic data collection to monitor epidemiological trends over time in a defined catchment population, and can use various indicators. Data can be obtained from reference laboratories, outbreak reports, hospital records or sentinel systems, and use consistent case definitions and reporting methods. There is usually no known population denominator, thus surveillance data cannot be used to measure disease incidence. Limitations include under-detection/under-reporting (if passive surveillance) or over-reporting (e.g., due to improvements in case detection or introduction of new vaccines with increased disease awareness). Changes/delays in case counting or reporting can artificially reduce the number of reported cases, thus artificially increasing vaccine effectiveness. Infectious Disease Surveillance (International Encyclopedia of Public Health 2017;222-9) is a comprehensive review including definitions, methods, and considerations on use of surveillance data in vaccine studies. The chapter on Routine Surveillance of Infectious Diseases in Modern Infectious Disease Epidemiology (J. Giesecke. 3rd Ed. CRC Press, 2017) discusses how surveillance data are collected and interpreted, and identifies sources of potential bias. Chapter 8 of Vaccination Programmes | Epidemiology, Monitoring, Evaluation outlines the main methods of vaccine-preventable disease surveillance, considering data sources, case definitions, biases and methods for descriptive analyses.
Granular epidemiological surveillance data (e.g., by age, gender, pathogen strain) are of particular importance for vaccine effectiveness studies. Such data were available from the European Centre for Disease Prevention and Control and the WHO Coronavirus (COVID-19) Dashboard during the COVID-19 pandemic and, importantly, also included vaccine coverage data.
EHRs and claims-based databases constitute an alternative to epidemiological surveillance data held by national public health bodies, as illustrated in Using EHR data to identify coronavirus infections in hospitalized patients: Impact of case definitions on disease surveillance (Int J Med Inform. 2022;166:104842), which also recommends using sensitivity analyses to assess the impact of variations in case definitions.
Examples of vaccination registries, and challenges in developing such registries, are discussed in Vaccine registers-experiences from Europe and elsewhere (Euro Surveill. 2012;17(17):20159), Validation of the new Swedish vaccination register - Accuracy and completeness of register data (Vaccine 2020; 38(25):4104-10), and Establishing and maintaining the National Vaccination Register in Finland (Euro Surveill. 2017;22(17):30520). Developed by WHO, Public health surveillance for COVID-19: interim guidance describes key aspects of the implementation of SARS-CoV-2 surveillance, including a section on vaccine effectiveness monitoring in relation to surveillance systems.
16.2.2.3. Study designs for vaccine effectiveness assessment
Traditional cohort and case-control designs
The case-control design has been used to evaluate vaccine effectiveness, but the likelihood of bias and confounding is a potential important limitation. The articles Case-control vaccine effectiveness studies: Preparation, design, and enrollment of cases and controls (Vaccine 2017; 35(25):3295-302) and Case-control vaccine effectiveness studies: Data collection, analysis and reporting results (Vaccine 2017; 35(25):3303-8) provide recommendations on best practices for their design, analysis and reporting. Based on a meta-analysis of 49 cohort studies and 10 case-control studies, Efficacy and effectiveness of influenza vaccines in elderly people: a systematic review (Lancet 2005;366(9492):1165-74) highlights the heterogeneity of outcomes and study populations included in such studies and the high likelihood of selection bias. In A Dynamic Model for Evaluation of the Bias of Influenza Vaccine Effectiveness Estimates From Observational Studies (Am J Epidemiol. 2019;188(2):451-60), a dynamic probability model was developed to evaluate biases in passive surveillance cohort, test-negative, and traditional case-control studies.
Non-specific effects of vaccines, such as a decrease of mortality, have been claimed in observational studies but can be affected by bias and confounding. Epidemiological studies of the 'non-specific effects' of vaccines: I--data collection in observational studies (Trop Med Int Health 2009;14(9):969-76.) and Epidemiological studies of the non-specific effects of vaccines: II--methodological issues in the design and analysis of cohort studies (Trop Med Int Health 2009;14(9):977-85) provide recommendations for observational studies conducted in high mortality settings; however, these recommendations have wider relevance.
The cohort design has been widely used to monitor the effectiveness of COVID-19 vaccines; the following two examples reflect early times of the pandemic, and its later phase when several vaccines were used, reaching wider population groups and used according to different types of vaccination schedule depending on national policies: BNT162b2 mRNA Covid-19 Vaccine in a Nationwide Mass Vaccination Setting (N Engl J Med. 2021;384(15):1412-23) used data from a nationwide healthcare organisation to match vaccinated and unvaccinated subjects according to demographic and clinical characteristics, to assess effectiveness against infection, COVID-19 related hospitalisation, severe illness, and death. Vaccine effectiveness against SARS-CoV-2 infection, hospitalization, and death when combining a first dose ChAdOx1 vaccine with a subsequent mRNA vaccine in Denmark: A nationwide population-based cohort study (PLoS Med. 2021;18(12):e1003874) used nationwide linked registries to estimate VE against several outcomes of interest of a heterologous vaccination schedule, compared to unvaccinated individuals. As vaccination coverage increased, using a non-vaccinated comparator group became no longer feasible or suitable, and alternative comparators were needed (see paragraph below on comparative effectiveness).
More recently, pharmacoepidemiological studies have assessed the effectiveness of COVID-19 booster vaccination, which uncovered new methodological challenges, such as the need to account for time-varying confounding. Challenges in Estimating the Effectiveness of COVID-19 Vaccination Using Observational Data (Ann Intern Med. 2023;176(5):685-693) describes two approaches to target trial emulation to overcome limitations due to confounding or designs not considering the evolution of the pandemic over time and the rapid uptake of vaccination. Comparative effectiveness of different primary vaccination courses on mRNA-based booster vaccines against SARs-COV-2 infections: a time-varying cohort analysis using trial emulation in the Virus Watch community cohort (Int J Epidemiol. 2023 Apr 19;52(2):342-354) conducted trial emulation by meta-analysing eight cohort results to reduce time-varying confounding-by-indication.
Test-negative case-control design
The test-negative case-control design aims to reduce bias associated with misclassification of infection and confounding by healthcare-seeking behaviour, at the cost of sometimes difficult-to-test assumptions. The test-negative design for estimating influenza vaccine effectiveness (Vaccine 2013;31(17):2165-8) explains the rationale, assumptions and analysis of this design, originally developed for influenza vaccines. Study subjects were all persons seeking care for an acute respiratory illness, and influenza VE was estimated from the ratio of the odds of vaccination among subjects testing positive for influenza to the odds of vaccination among subject testing negative. Test-Negative Designs: Differences and Commonalities with Other Case-Control Studies with "Other Patient" Controls (Epidemiology. 2019 Nov;30(6):838-44) discusses advantages and disadvantages of the design in various circumstances. The use of test-negative controls to monitor vaccine effectiveness: a systematic review of methodology (Epidemiology 2020;31(1):43-64) discusses challenges of this design for various vaccines and pathogens, also providing a list of recommendations.
In Effectiveness of rotavirus vaccines in preventing cases and hospitalizations due to rotavirus gastroenteritis in Navarre, Spain (Vaccine 2012;30(3):539-43), electronic clinical reports were used to select cases (children with confirmed rotavirus infection) and test-negative controls (children who tested negative for rotavirus in all samples), under the assumption that the rate of gastroenteritis caused by pathogens other than rotavirus is the same in both vaccinated and unvaccinated subjects. A limitation is sensitivity of the laboratory test, which may underestimate vaccine effectiveness. In addition, if the viral type is not available, it is not possible to study the association between vaccine failure and a possible mismatch between vaccine strains and circulating strains. These learnings still apply today in the context of COVId-19 vaccines.
The article Theoretical basis of the test-negative study design for assessment of influenza vaccine effectiveness (Am J Epidemiol. 2016;184(5):345-53; see also the related Comments) uses directed acyclic graphs to characterise potential biases and shows how they can be avoided or minimised. In Estimands and Estimation of COVID-19 Vaccine Effectiveness Under the Test-Negative Design: Connections to Causal Inference (Epidemiology 2022;33(3):325-33), an unbiased estimator for vaccine effectiveness using the test-negative design is proposed under the scenario of different vaccine effectiveness estimates across patient subgroups.
In the multicentre study in 18 hospitals 2012/13 influenza vaccine effectiveness against hospitalised influenza A(H1N1)pdm09, A(H3N2) and B: estimates from a European network of hospitals (EuroSurveill 2015;20(2):pii=21011), influenza VE was estimated based on the assumption that confounding due to health-seeking behaviour is minimised since all individuals needing hospitalisation are likely to be hospitalised.
Postlicensure Evaluation of COVID-19 Vaccines (JAMA. 2020;324(19):1939-40) describes methodological challenges of the test-negative design applied to COVID-19 vaccines and discusses solutions to minimise bias. Covid-19 Vaccine Effectiveness and the Test-Negative Design (N Engl J Med. 2021;385(15):1431-33) uses the example of a published study in a large hospital network to provide considerations on how to report findings and assess their sensitivity to biases specific to the test-negative design. The study Effectiveness of the Pfizer-BioNTech and Oxford-AstraZeneca vaccines on covid-19 related symptoms, hospital admissions, and mortality in older adults in England: test negative case-control study (BMJ 2021;373:n1088) linked routine community testing and vaccination data to estimate effectiveness against confirmed symptomatic infection, COVID-19 related hospital admissions and case fatality, and estimated the odds ratios for testing positive to SARS-CoV-2 in vaccinated compared to unvaccinated subjects with compatible symptoms. The study also provides considerations on strengths and limitations of the test-negative design.
Case-population, case-coverage, and screening methods
These methods are related, and all include, to some extent, an ecological component such as vaccine coverage or epidemiological surveillance data at population level. Terms to refer to these designs are sometimes used interchangeably. The case-coverage design is discussed above in paragraph 16.2.2.2. Case-population studies are described in Chapter 4.2.5 and in Vaccine Case-Population: A New Method for Vaccine Safety Surveillance (Drug Saf. 2016;39(12):1197-209).
The screening method estimates vaccine effectiveness by comparing vaccination coverage in positive (usually laboratory confirmed) cases of a disease (e.g., influenza) with the vaccination coverage in the population from which the cases are derived (e.g., in the same age group). If representative data on cases and vaccination coverage are available, it can provide an inexpensive and rapid method to provide early estimates or identify changes in effectiveness over time. However, Application of the screening method to monitor influenza vaccine effectiveness among the elderly in Germany (BMC Infect Dis. 2015;15(1):137) emphasises that accurate and age-specific vaccine coverage data are crucial to provide valid estimates. Since adjusting for important confounders and assessing product-specific effectiveness is generally challenging, this method should be considered mainly as a supplementary tool to assess crude effectiveness. COVID-19 vaccine effectiveness estimation using the screening method – operational tool for countries (2022) also provides a good introduction to the method and its strengths and limitations.
Indirect cohort (Broome) method
The indirect cohort method is a case-control type design which uses cases caused by non-vaccine serotypes as controls, and uses surveillance data, instead of vaccination coverage data. Use of surveillance data to estimate the effectiveness of the 7-valent conjugate pneumococcal vaccine in children less than 5 years of age over a 9 year period (Vaccine 2012;30(27):4067-72) evaluated the effectiveness of a pneumococcal conjugate vaccine against invasive pneumococcal disease and compared to the results of a standard case-control design conducted during the same time period. The authors consider the method most useful shortly after vaccine introduction, and less useful in a setting of very high vaccine coverage and fewer cases. Using the indirect cohort design to estimate the effectiveness of the seven valent pneumococcal conjugate vaccine in England and Wales (PLoS One 2011;6(12):e28435) and Effectiveness of the seven-valent and thirteen-valent pneumococcal conjugate vaccines in England: The indirect cohort design, 2006-2018 (Vaccine 2019;37(32):4491-98) describe how the method was used to estimate effectiveness of various vaccine schedules as well as for each vaccine serotype.
Density case-control design
Effectiveness of live-attenuated Japanese encephalitis vaccine (SA14-14-2): a case-control study (Lancet 1996;347(9015):1583-6) describes a case-control study of incident cases in which the control group consisted of all village-matched children of a given age who were at risk of developing disease at the time that the case occurred (density sampling). The effect measured is an incidence density rate ratio. In Vaccine Effectiveness of Polysaccharide Vaccines Against Clinical Meningitis - Niamey, Niger, June 2015 (PLoS Curr. 2016;8), a case-control study compared the odds of vaccination among suspected meningitis cases to controls enrolled in a vaccine coverage survey performed at the end of the epidemic. A simulated density case-control design randomly attributing recruitment dates to controls based on case dates of onset was used to compute vaccine effectiveness. In Surveillance of COVID-19 vaccine effectiveness: a real-time case–control study in southern Sweden (Epidemiol Infect. 2022;150:1-15) a continuous density case-control sampling was performed, with the control group randomly selected from the complete study cohort as individuals without a positive test the same week as the case or 12 weeks prior.
Waning immunity
Studying how immunity conferred by vaccination wanes over time requires consideration of within-host dynamics of the pathogen and immune system, as well as the associated population-level transmission dynamics. Implications of vaccination and waning immunity (Proc Biol Sci. 2009; 276(1664):2071-80) combined immunological and epidemiological models of measles infection to examine the interplay between disease incidence, waning immunity and boosting.
Global Varicella Vaccine Effectiveness: A Meta-analysis (Pediatrics 2016; 137(3):e20153741) highlights the challenges to reliably measure effectiveness when some confounders cannot be controlled for, force of infection may be high, degree of exposure in study participants may be variable, and data may originate from settings where there is evidence of vaccine failure. Several estimates or studies may therefore be needed to accurately conclude in waning immunity. Duration of effectiveness of vaccines against SARS-CoV-2 infection and COVID-19 disease: results of a systematic review and meta-regression (Lancet 2022;399(10328):924-944) reviews evidence of changes in efficacy or effectiveness with time since full vaccination for various clinical outcomes; biases in evaluating changes in effectiveness over time, and how to minimise them, are presented in a tabular format. Effectiveness of Covid-19 Vaccines over a 9-Month Period in North Carolina (N Engl J Med. 2022;386(10):933-941) linked COVID-19 surveillance and vaccination data to estimate reduction in current risks of infection, hospitalisation and death as a function of time elapsed since vaccination, and demonstrated durable effectiveness against hospitalisation and death while waning protection against infection over time was shown to be due to both declining immunity and emergence of the delta variant.
Vaccine effectiveness estimates over time are subject to bias from differential depletion of susceptibles (persons at risk of infection) between vaccinated and unvaccinated groups, which can lead to biased estimates of waning effectiveness. Depletion-of-susceptibles bias in influenza vaccine waning studies: how to ensure robust results (Epidemiol Infect. 2019;147:e306) recommends to study only vaccinated persons, and compare for each day the incidence in persons with earlier or later dates of vaccination, to assess waning as a function of vaccination time. Identifying and Alleviating Bias Due to Differential Depletion of Susceptible People in Postmarketing Evaluations of COVID-19 Vaccines (Am J Epidemiol. 2022;191(5):800-11) outlines scenarios under which bias can arise and identifies approaches to minimise these biases.
Comparative vaccine effectiveness
Comparing vaccine benefits has traditionally been performed using head-to-head immunogenicity studies, while comparative effectiveness designs have been used mostly to compare vaccination schedules, vaccine formulations, or administration routes (e.g., for measles, mumps and rubella (MMR), influenza, or pneumococcal vaccines; see for example, Analysis of relative effectiveness of high-dose versus standard-dose influenza vaccines using an instrumental variable method (Vaccine 2019;37(11):1484-90). Methods to account for measured and unmeasured confounders in influenza relative vaccine effectiveness studies: A brief review of the literature (Influenza Other Respir. Viruses 2022;16(5):846-850) discusses methods to account for confounding in such studies. In The risk of non-specific hospitalised infections following MMR vaccination given with and without inactivated vaccines in the second year of life. Comparative self-controlled case-series study in England (Vaccine 2019;37(36):5211-17) the SCCS design was used to compare the effectiveness of the MMR vaccine alone with the MMR vaccine in combination with PCV7 or with both PCV7 and the combined Hib-MenC vaccine. Comparative effectiveness of pneumococcal vaccination with PPV23 and PCV13 in COPD patients over a 5-year follow-up cohort study, (Sci Rep 2021;11(1):15948.) used a prospective cohort design to compare effectiveness between the 23-valent vaccine, the 13-valent vaccine, and no vaccination.
The COVID-19 vaccination campaigns increased the interest in, and triggered, comparative effectiveness studies. Postmarketing studies: can they provide a safety net for COVID-19 vaccines in the UK? (BMJ Evid Based Med. 2020:bmjebm-2020-111507) discusses methodological and operational aspects and provides considerations on head-to-head vaccine comparisons. Assessment of Effectiveness of 1 Dose of BNT162b2 Vaccine for SARS-CoV-2 Infection 13 to 24 Days After Immunization (JAMA Netw Open. 2021;4(6):e2115985) compared the effectiveness of the first vaccine dose between two post-immunisation periods. Comparative effectiveness of the BNT162b2 and ChAdOx1 vaccines against Covid-19 in people over 50 (Nat Commun. 2022;13(1):1519) used data from the UK Biobank linked to primary care, hospital admissions, and COVID-19 testing data, to compare the effectiveness of BNT162b2 vs. ChAdOx1s against COVID-19 infection and hospitalisation, using propensity score modelling. Comparative Effectiveness of BNT162b2 and mRNA-1273 Vaccines in U.S. Veterans (N Engl J Med. 2022;386(2):105-15) and Comparative effectiveness of BNT162b2 versus mRNA-1273 covid-19 vaccine boosting in England: matched cohort study in OpenSAFELY-TPP used a target trial emulation design.
Comparative vaccine effectiveness studies may require larger sample sizes, as they aim to detect smaller effect sizes as opposed to effectiveness studies for a single vaccine, where an unvaccinated group is used as a comparator. Various sources of confounding (such as self-seeking testing behaviour) should be considered, and appropriate methods used, such as (propensity score) matching, instrumental variable analysis, inverse probability of treatment weighting, use of negative control outcomes, off-season outcomes (for influenza vaccines) and positive control outcomes. For some vaccines (e.g., COVID-19 vaccines), variant-specific comparative effectiveness data are important, taking into consideration the correlation between vaccine schedules and calendar periods, and therefore with variants in circulation at a given time.
Impact studies
Vaccine impact studies estimate disease reduction in a community. These studies are typically ecological or modelling analyses that compare disease outcomes pre- and post-vaccine introduction. Reductions in disease outcomes are observed through direct effects of vaccination in vaccinated people, and indirect effects due to reduced transmission within a community. Other concurrent interventions or phenomena unrelated to vaccine effects, such as changes in risk behaviours or healthcare practices, may reduce disease outcomes and confound the assessment of vaccine impact (see The value of vaccine programme impact monitoring during the COVID-19 pandemic, Lancet 2022;399(10320):119-21). For example, for a paediatric vaccine, the impact of vaccination can be quantified in the targeted age group (overall effect) or in other age groups (indirect effect). For an overview, see Vaccine effects and impact of vaccination programmes in post-licensure studies (Vaccine 2013;31(48):5634-42).
Direct and indirect effects in vaccine efficacy and effectiveness (Am J Epidemiol. 1991;133(4):323-31) describes how parameters intended to measure direct effects must be robust and interpretable in the midst of complex indirect effects of vaccine intervention programmes. Lack of impact of rotavirus vaccination on childhood seizure hospitalizations in England - An interrupted time series analysis (Vaccine 2018; 36(31):4589-92) discusses possible reasons for negative findings compared to previous studies. In a review of 65 articles, Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: updated systematic review and meta-analysis (Lancet. 2019;394(10197):497–509) compared the prevalence or incidence of several HPV-related endpoints between the pre- and post-vaccination periods with stratification by sex, age, and years since introduction of HPV vaccination.
Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: an observational study using national surveillance data (Lancet. 2021;397(10287):1819-29) evaluated the public health impact of vaccination using national surveillance and vaccine uptake data. Although such population-level data are ecological, and teasing apart the impact of the vaccination programme from the impact of non-pharmaceutical interventions is complex, declines in incident cases by age group were shown to be aligned with high vaccine coverage rather than initiation of the nationwide lockdown.
Accumulated effectiveness data has suggested the potential for a population-level effect of COVID-19 vaccination, which has been critical to control the pandemic. Community-level evidence for SARS-CoV-2 vaccine protection of unvaccinated individuals (Nat Med. 2021;27(8):1367-9) analysed vaccination records and test results in a large population, while mitigating the confounding effect of natural immunity and the spatiotemporally dynamic nature of the epidemic, and showed that vaccination provided cross-protection to unvaccinated individuals in the community.
Transmission studies
Vaccination programmes have indirect effects at the population-level, also called herd immunity, as a result of reduced transmission. Besides measuring the direct effect of vaccination in vaccine effectiveness studies, it is important to assess whether vaccination has an effect on transmission. As a high-risk setting, households can provide evidence of such impact.
Among the first studies of the impact of COVID-19 vaccination on transmission, Effect of Vaccination on Household Transmission of SARS-CoV-2 in England (N Engl J Med. 2021;385(8):759-60) was a nested case-control study estimating odds ratios for household members becoming secondary cases if the case was vaccinated within 21 days or more before testing positive, vs. household members where the case was not vaccinated. Vaccination with BNT162b2 reduces transmission of SARS-CoV-2 to household contacts in Israel (Science. 2022;375(6585):1151-54) assessed the effectiveness of BNT162b2 against susceptibility to infection and infectiousness, comparing pre- and post-Delta periods, using a chain binomial model applied to data from a large healthcare organisation. Community transmission and viral load kinetics of the SARS-CoV-2 delta (B.1.617.2) variant in vaccinated and unvaccinated individuals in the UK: a prospective, longitudinal, cohort study (Lancet Infect Dis. 2022;22(2):183-95) ascertained secondary transmission by longitudinally following index cases and their contacts (regardless of symptoms) early after exposure to the Delta variant, and highlights the importance of community studies to characterise transmission in highly vaccinated populations.
Specific limitations of transmission studies such as likelihood of information bias (misclassification) and selection bias, should be considered when interpreting findings and are discussed in the above references.
Cluster design
A cluster is a group of subjects sharing common characteristics: geographical (community, administrative area), health-related (hospital), educational (schools), or social (household). In cluster randomised trials, clusters instead of individual subjects are randomly allocated to an intervention, whereas in infectious disease epidemiology studies, clusters are sampled based on aspects of transmission (e.g., within a community) or a vaccination programme. This design is often used in low and middle income settings and can measure vaccination interventions naturally applied at the cluster level or when the study objectives require a cluster design (e.g., to estimate herd immunity).
Meningococcal B Vaccine and Meningococcal Carriage in Adolescents in Australia (N Engl J Med. 2020;382(4):318-27) used cluster randomisation to assign students, according to school, to receive 4CMenB vaccination, either at baseline or at 12 months (as a control) to measure oropharyngeal carriage.
In The ring vaccination trial: a novel cluster randomised controlled trial design to evaluate vaccine efficacy and effectiveness during outbreaks, with special reference to Ebola (BMJ. 2015;351:h3740), a newly diagnosed Ebola case served as the index case to form a “ring”, which was then randomised to immediate or delayed vaccination with inclusion based on tracing cases using active surveillance instead of randomisation. In Assessing the safety, impact and effectiveness of RTS,S/AS01 E malaria vaccine following its introduction in three sub-Saharan African countries: methodological approaches and study set-up (Malar J. 2022;21(1):132), active surveillance was used to enrol large numbers of children in vaccinated and unvaccinated clusters as part of the WHO Malaria Vaccine Implementation Programme, to conduct temporal (before vs. after) and concurrent (exposed vs. unexposed) cluster comparisons. Clusters were selected based on geographically limited areas with demographic surveillance in place and infrastructure to monitor population health and vaccination programmes.
Misclassification in studies of vaccine effectiveness
Like vaccine safety studies, studies of vaccine effectiveness rely on accurate identification of vaccine exposure and of cases of the targeted vaccine-preventable disease/infection, but in practice, diagnostic tests, clinical case definitions and vaccination records often present inaccuracies. Bias due to differential and non-differential disease- and exposure misclassification in studies of vaccine effectiveness (PLoS One 2018;15;13(6):e0199180) explores through simulations the impact of non-differential and differential disease- and exposure-misclassification when estimating vaccine effectiveness using cohort, case-control, test-negative case-control and case-cohort designs. This can also be applied to safety outcomes, especially those with a complex natural history such as neurological or potential immune mediated diseases, and is particularly relevant for secondary use of data, where validation studies may be needed in a first step. Misclassification can lead to significant bias and its impact strongly depends on the vaccination scenarios. A web application designed in the ADVANCE project is publicly available to assess the potential (joint) impact of possibly differential disease- and exposure misclassification.
16.2.3. Specific aspects of pharmacoepidemiological vaccine studies
16.2.3.1. Studies in special populations
Special populations include pregnant and breastfeeding persons, immunocompromised patients (including transplanted patients), paediatric populations, older adults/the elderly, and patients with rare disorders. Post-authorisation studies are often required for these populations, which are usually not included in the clinical development of vaccines. In real-world settings, special populations are often the subject of specific vaccination recommendations, which may impact study designs and choice of an appropriate comparator. This was the case, for example, of COVID-19 vaccines which initially targeted high-risk priority groups. The article Vaccine safety in special populations (Hum Vaccin. 2011;7(2):269-71) highlights design issues when evaluating vaccine safety in these populations. Methodological challenges include defining the study population (particularly for immunocompromised populations), low sample size due to rare outcomes, accounting for comorbidities and other types of confounders, or difficulty in identifying cases or disease duration and severity in immunocompromised patients.
Influenza vaccination for immunocompromised patients: systematic review and meta-analysis by etiology (J Infect Dis. 2012;206(8):1250-9) illustrates the importance of performing stratified analyses by aetiology of immunocompromised status and limitations due to residual confounding, differences within and between etiological groups and small sample size in some subgroups. In anticipation of the design of post-authorisation vaccine effectiveness and safety studies, the study Burden of herpes zoster in 16 selected immunocompromised populations in England: a cohort study in the Clinical Practice Research Datalink 2000–2012 (BMJ Open 2018;8(6): e020528) illustrated the challenges of defining an immunocompromised cohort and a relevant comparator cohort in a primary healthcare database. Validation of a Method to Identify Immunocompromised Patients with Severe Sepsis in Administrative Databases (Ann Am Thorac Soc. 2016;13(2):253-8) provides considerations on identifying this group of patients in large administrative databases.
Pregnant and breastfeeding persons represent an important group to be addressed when monitoring vaccine use; Annex 2 of this Guide provides guidance on methods to evaluate medicines in pregnancy and breastfeeding, including for vaccine studies. The Guidance for design and analysis of observational studies of foetal and newborn outcomes following COVID-19 vaccination during pregnancy (Vaccine 2021;39(14):1882-6) provides useful insights on study design, data collection, and analytical issues in COVID-19 vaccine safety studies in pregnant people, and can be applied to other vaccines.
16.2.3.2. Meta-analyses
The guidance on conducting meta-analyses of pharmacoepidemiological studies of safety outcomes (Annex 1 of this Guide) is also applicable to vaccines. A systematic review evaluating the potential for bias and the methodological quality of meta-analyses in vaccinology (Vaccine 2007;25(52):8794-806) provides a comprehensive overview of quality and limitations of meta-analyses. Meta-analysis of the risk of autoimmune thyroiditis, Guillain-Barré syndrome, and inflammatory bowel disease following vaccination with AS04-adjuvanted human papillomavirus 16/18 vaccine (Pharmacoepidemiol Drug Saf. 2020;29(9):1159-67) combined data from 18 randomised controlled trials, one cluster-randomised trial, two large observational retrospective cohort studies, and one case-control study, resulting in a large sample size for these rare events. The Systematic review and meta-analysis of the effectiveness and perinatal outcomes of COVID-19 vaccination in pregnancy (Nat Commun. 2022;13(1):2414) generated evidence on a large number of adverse pregnancy and perinatal outcomes.
Meta-analytical methods are increasingly used in multi-database studies (see Chapter 9) to combine data generated at country level to obtain pooled risk estimates in large populations. In SARS-CoV-2 Vaccination and Myocarditis in a Nordic Cohort Study of 23 Million Residents (JAMA Cardiol. 2022;7(6):600-12), four cohort studies were conducted in linked nationwide health registers in Denmark, Finland, Norway, and Sweden according to a common protocol; the results were combined using meta-analysis and the homogeneity of country-specific estimates was tested.
16.2.3.3. Pharmacogenetic studies
There is increasing interest in the role of genomics in pharmacoepidemiology (see Chapter 16.3), including for the study of vaccine safety outcomes (see Adversomics: a new paradigm for vaccine safety and design, Expert Rev Vaccines 2015; 14(7): 935–47). Vaccinomics and Adversomics in the Era of Precision Medicine: A Review Based on HBV, MMR, HPV, and COVID-19 Vaccines (J Clin Med. 2020;9(11):3561) highlights that knowledge of genetic factors modulating responses to vaccination could contribute to the evaluation of vaccine safety and effectiveness. In State-wide genomic epidemiology investigations of COVID-19 in healthcare workers in 2020 Victoria, Australia: Qualitative thematic analysis to provide insights for future pandemic preparedness (Lancet Reg Health West Pac. 2022;25:100487), a large SARS-CoV-2 genomic epidemiological investigation identified transmission dynamics using a newly developed set of metadata. Genetic risk and incident venous thromboembolism in middle-aged and older adults following COVID-19 vaccination (J Thromb Haemost. 2022;20(12):2887-2895) used data from the UK Biobank to estimate hazard ratios of the associations between a polygenic risk score and post-vaccination incident veinous thromboembolism.
16.2.3.4. Generic protocols
Generic protocols, also referred to as template or master protocols, provide a standardised structure to support study design and protocol development. Such protocols have supported the urgent need for COVID-19 vaccine monitoring, often based, in Europe, on the EMA Guidance for the format and content of the protocol of non-interventional post-authorisation safety studies (2012).
A protocol for generating background rates of AESIs for the monitoring of COVID-19 vaccines (2021) was developed by the vACcine Covid-19 monitoring readinESS (ACCESS) consortium, which also published Template study protocols (2021) to support the design of safety studies, based on both cohort-event monitoring and secondary use of data. The protocol Rapid assessment of COVID-19 vaccines safety concerns through electronic health records- a protocol template from the ACCESS project compares the suitability of the ecological design and the unadjusted self-controlled risk interval (SCRI) for rapid safety assessment, by type of AESI. Other published templates include FDA’s Background Rates of Adverse Events of Special Interest for COVID-19 Vaccine Safety Monitoring protocol, the COVID-19 Vaccine Safety Active Monitoring Protocol and the Master Protocol: Assessment of Risk of Safety Outcomes Following COVID-19 Vaccination (FDA BEST Initiative, 2021); and the Template for observational study protocols for sentinel surveillance of adverse events of special interest (AESIs) after vaccination with COVID-19 vaccines (WHO, 2021).
The ACCESS consortium also published template protocols (2021) for COVID-19 vaccine effectiveness studies using the cohort and test-negative case-control designs. The Core protocol for ECDC studies of COVID-19 vaccine effectiveness against hospitalisation with Severe Acute Respiratory Infection laboratory-confirmed with SARS-CoV-2 (ECDC, 2021) presents the main elements to consider to design multi-centre, multi-country hospital-based COVID-19 vaccine effectiveness studies in patients hospitalised with severe acute respiratory infections (SARI).
The DRIVE project developed a Core protocol for type/brand specific influenza vaccine effectiveness studies - Test-negative design studies and a Core protocol for population-based database cohort-studies, and the COVIDRIVE consortium a Brand-specific COVID-19 vaccine effectiveness protocol to assess effectiveness against severe COVID-19 disease.
Generic protocols for retrospective case-control studies and retrospective cohort studies to assess the effectiveness of rotavirus and influenza vaccination in EU Member States are published by ECDC and describe potential data sources to identify virological outcomes. The Protocol for Cluster Investigations to Measure Influenza Vaccine Effectiveness (ECDC, 2009) builds on the cluster design to generate rapid/early influenza season estimates in settings where investigation can take place at the same time as vaccination is carried out (e.g. schools, care homes). The generic study protocol to assess the impact of rotavirus vaccination (ECDC, 2013) lists the information to be collected to compare the incidence/proportion of rotavirus cases in the period before and after vaccine introduction.
Although developed for specific vaccines, all these protocols can be tailored to other vaccine exposures and outcomes, as they address the most important aspects to consider for the design of vaccine safety and effectiveness studies.
16.3. Design, implementation and analysis of pharmacogenetic studies
16.3.1. Introduction
Individual differences in the response to medicines encompass variation in both efficacy/effectiveness and safety, including the risk of severe adverse drug reactions. Clinical factors influencing response include disease severity, age, gender, and concomitant drug use. However, natural genetic variation that influences the expression or activity of proteins involved in drug disposition (absorption, metabolism, distribution, and excretion) as well as the protein targets of drug action (such as enzymes and receptors) may be an important additional source of inter-individual variability in both the beneficial and adverse effects of drugs (see Pharmacogenomics: translating functional genomics into rational therapeutics. Science 1999;286(5439):487-91).
Pharmacogenetics is defined as the study of variation in the DNA sequence as a determinant of drug response. Drug response may vary as a result of differences in the DNA sequence present in the germline or, in the case of cancer treatments, due to somatic variation in the DNA arising in cancer cells (see The Roles of Common Variation and Somatic Mutation in Cancer Pharmacogenomics, Oncol Ther. 2019;7(1):1-32; Systematic pan-cancer analysis of mutation-treatment interactions using large real-world clinicogenomics data, Nat Med. 2022 Aug;28(8):1656-1661). Notably, in the case of treatment or prevention of infectious diseases, the genome of both the pathogen and the host may influence drug and vaccine responses, either independently, interactively or jointly (see Pharmacogenomics and infectious diseases: impact on drug response and applications to disease management, Am J Health Syst Pharm. 2002;59(17):1626-31; The potential of genomics for infectious disease forecasting, Nat Microbiol. 2022 Nov;7(11):1736-1743). For example, COVID-19 vaccine effectiveness changes significantly according to SARS-CoV-2 variant, likely due to vaccine-escape mutations in the virus genome (see Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries, Genomics 2021; 113(4):2158-2170 and Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant, N Engl J Med 2022; 386:1532-46). When incorporated, the study of genetic variation underlying drug response can complement information on clinical factors and disease sub-phenotypes to optimise the prediction of treatment response and reduce the risk of adverse reactions. The identification of variation in genes that modify the response to drugs provides an opportunity to optimise safety and effectiveness of the currently available drugs and to develop new drugs for paediatric and adult populations (see Drug discovery: a historical perspective, Science 2000;287(5460):1960-4).
The broader term pharmacogenomics has been used to describe the study changes both in the DNA and RNA, and how they may determine drug response. However, the distinction between pharmacogenetics and pharmacogenomics is arbitrary, and both terms are used interchangeably. It is important to note that pharmacogenomics is one of several approaches available to identify useful biomarkers of drug effects. Other approaches include, but are not limited to, epigenomics (the study of gene expression changes not attributable to changes in the DNA sequence), transcriptomics, proteomics (protein function and levels, see Precision medicine: from pharmacogenomics to pharmacoproteomics, Clin Proteom. 2016; 13:25), and metabolomics.
16.3.2. Identification of genetic variants influencing drug response
Approaches
Identification of genetic variation associated with important drug or therapy-related outcomes can be carried out by three main technologies. Their choice may be dictated by whether the aim is research and discovery or clinical application, and whether the genetic variants being sought occur at high or low frequency in the population or patient group(s) being evaluated. The strategy to identify genetic variants will depend on the aim and design of the pharmacogenetic study or the clinical application (see Methodological and statistical issues in pharmacogenomics, J Pharm Pharmacol. 2010;62(2):161-6). For illustration, to assess clinical applications, technologies might be used to identify genetic variants where there is already prior knowledge about the gene or the variant (candidate gene studies). These studies require prior information about the likelihood of the polymorphism, gene, or gene-product interacting with a drug or drug pathway, and thus, resources can be directed to several important genetic polymorphisms with a higher a priori chance of relevant drug-gene interactions. Moving towards individualized medicine with pharmacogenomics (Nature 2004;429(6990):464-8) explains that lack or incompleteness of information on genes from previous studies may result in the failure in identifying every important genetic determinant in the genome.
In contrast, genome-wide scan approaches are discovery orientated and use technologies that identify genetic variants across the genome without previous information or gene/variant hypothesis (hypothesis-generating or hypothesis-agnostic approach). Genome-wide approaches are widely used to discover the genetic basis of common complex diseases where multiple genetic variations contribute to disease risk. The same study design is applicable to identification of genetic variants that influence treatment response. However, common variants in the genome, if functional, have generally small effect sizes, and therefore large sample sizes should be considered, for example by pooling different studies as done by the CHARGE Consortium with its focus on cardiovascular diseases (see The Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium as a model of collaborative science, Epidemiology 2013;24(3):346-8). By comparing the frequency of genetic variants between drug responders and non-responders, or those with or without drug toxicity, genome-wide approaches can identify important genetic determinants. They may detect variants in genes, which were previously not considered as candidate genes, or even variants outside of the genes. However, because of the concept of linkage disequilibrium, whereby certain genetic determinants tend to be co-inherited together, it is possible that the genetic associations identified through a genome-wide approach may not be truly biologically functional polymorphisms, but instead may simply be a linkage-related marker of another genetic determinant that is the true biologically relevant genetic determinant. Thus, this approach is considered discovery in nature. Furthermore, failure to cover all relevant genetic risk factors can still be a problem, though less than with the candidate gene approach. It is therefore essential to conduct replication studies in independent cohorts and validation studies (in vivo and in vitro) to ascertain the generalisability of findings to populations of individuals, to characterise the mechanistic basis of the effect of these genes on drug action, and to identify true biologic genetic determinants. Importantly, allele frequencies differ across populations, and these differences should be accounted for to reduce biases when designing and analysing pharmacogenetic studies, and to ensure equity when implementing pharmacogenomics in the healthcare setting (see Preventing the exacerbation of health disparities by iatrogenic pharmacogenomic applications: lessons from warfarin, Pharmacogenomics 2018 19(11):875-81).
More recently, pharmacogenomic studies have also been performed in large national biobanks which link genetic data to healthcare data for cohorts of hundreds of thousands of subjects, such as the UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age (PLoS Med. 2015;12(3):e1001779) and the Estonian Biobank (see Cohort Profile: Estonian Biobank of the Estonian Genome Center, University of Tartu, Int J Epidemiol. 2015;44(4):1137-47). Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: challenges and solutions and other studies (Genet Med. 2019;21(6):1345-54) shows that these large-scale resources represent unique opportunities to discover novel and rare variants.
Technologies used for detection of genetic variants
The main technologies are:
-
Genotyping and array-based technologies which are the most feasible and cost-effective approach for most large-scale clinical utility studies and for clinical implementation, either through commercial or customised arrays. They can identify hundreds of thousands of genetic variants within one or several genes, including a common form of variations known as single nucleotide polymorphisms (SNPs). The identification of genetic determinants is limited to the variants included in the array, and thus, it cannot be used to discover novel variants. Generally, they are chosen on the grounds of biological plausibility, which may have been proven before in previous studies, or of knowledge of functional genes known to be involved in pharmacokinetic and pharmacodynamics pathways or related to the disease or intermediate phenotype.
-
Sanger sequencing represents the gold standard used in clinical settings for confirming genetic variants since it was first commercialised in 1986. More recently, Sanger sequencing has been replaced by other sequencing methods to increase the speed and reduce the cost of DNA sequencing, especially for automated analysis involving large numbers of samples.
-
Next generation sequencing (NGS) is a high-throughput sequencing technology that identifies genetic variants across the genome (whole genome sequencing; WGS) or the exome (whole exome sequencing; WES) without requiring prior knowledge on genetic biomarkers. These techniques may prove valuable in early research settings for discovery of novel or rare variants, and for the detection of structural variants and copy number variation which are common in pharmacogenes such as CYP2D6 (see A Review of the Important Role of CYP2D6 in Pharmacogenomics, Genes (Basel) 2020;11(11):1295; Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2C19 and CYP2D6 and SSRIs, Eur J Hum Genet. 2022 Oct;30(10):1114-1120; Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2C19 Genotype and Clopidogrel Therapy: 2022 Update, Clin Pharmacol Ther. 2022 Nov;112(5):959-967). As use of clinical WGS testing increases, the return of secondary pharmacogenomic findings will benefit from greater understanding of rare and novel variants.
Variant curation and annotation
Lastly, the identification of genetic variants requires careful curation and annotation to ensure that their description and allelic designation is standardised. Common pharmacogenomic variants and haplotypes (combinations of sequence variants in the same individual) are catalogued by the Pharmacogene Variation Consortium (PharmVar) using a ‘star allele’ nomenclature. The use of this nomenclature is historic and in human disease genetics, the reference sequence identifier (rs-id) is more commonly used as to assign a genetic variant unambiguously. Although the star allele nomenclature remains the most widely used classification in pharmacogenomic research it is recognised to have several limitations. Pharmacogenomic haplotypes and star alleles can lack accurate definition and validation, and there may be limited annotation of phenotypic effects. In addition, current classifications also exclude many rare variants which are increasingly recognised as having an important effect, as described in Pharmacogenetics at Scale: An Analysis of the UK Biobank (Clin Pharmacol Ther. 2021;109(6):1528-37). Some authors have called for an effort to standardise annotation sequence variants (see The Star-Allele Nomenclature: Retooling for Translational Genomics, Clin Pharmacol Ther. 2007;82(3):244–8).
16.3.3. Study designs
Several options are available for the design of pharmacogenetic studies to ascertain the effect and importantly the clinical relevance and utility of obtaining pharmacogenetic information to guide prescribing decisions regarding the choice and dose of agent for a particular condition (see Prognosis research strategy (PROGRESS) 4: Stratified medicine research, BMJ. 2013;346:e5793).
RCTs, both pre- and post-authorisation, provide the opportunity to address several pharmacogenetic questions. Pharmacogenetics in randomized controlled trials: considerations for trial design (Pharmacogenomics 2011;12(10):1485-92) describes three different trial designs differing in the timing of randomization and genotyping, and Promises and challenges of pharmacogenetics: an overview of study design, methodological and statistical issues (JRSM Cardiovasc Dis. 2012;1(1)) discusses outstanding methodological and statistical issues that may lead to heterogeneity among reported pharmacogenetic studies and how they may be addressed. Pharmacogenetic trials can be designed (or post hoc analysed) with the intention to study whether a subgroup of patients, defined by certain genetic characteristics, respond differently to the treatment under study. Alternatively, a trial can verify whether genotype-guided treatment is beneficial over standard care. Obvious limitations with regard to the assessment of rare adverse drug events or low prevalence genetic variants are the large sample size required and its related high costs. In order to make a trial as efficient as possible in terms of time, money and/or sample size, it is possible to opt for an adaptive trial design, which allows prospectively planned modifications in design after patients have been enrolled in the study. Such a design uses accumulating data to decide how to modify aspects of the study during its progress, without undermining the validity and integrity of the trial. An additional benefit is that the expected number of patients exposed to an inferior/harmful treatment can be reduced (see Potential of adaptive clinical trial designs in pharmacogenetic research, Pharmacogenomics 2012;13(5):571-8).
Observational studies are an alternative and can be family-based (using twins or siblings) or population-based (using unrelated individuals). The main advantage of family-based studies is the avoidance of bias due to population stratification. A clear practical disadvantage for pharmacogenetic studies is the requirement to study families where patients have been treated with the same drugs (see Methodological quality of pharmacogenetic studies: issues of concern, Stat Med. 2008;27(30):6547-69).
Population-based studies may be designed to assess drug-gene interactions as cohort (including exposure-only), case-cohort and case-control studies (including case-only, as described in Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls! Am J Epidemiol. 1996;144(3):207-13). Sound pharmacoepidemiological principles as described in this Guide also apply to observational pharmacogenetic studies. A specific type of confounding due to population stratification needs to be considered in pharmacogenetic studies, and, if present, needs to be dealt with. Its presence may be obvious where the study population includes more than one immediately recognisable ethnic group; however, in other studies stratification may be more subtle. Population stratification can be detected by the Pritchard and Rosenberg’s method, which involves genotyping additional SNPs in other areas of the genome and testing for association between them and outcome (see Association mapping in structured populations, Am J Hum Genet. 2000;67(1):170-81). In genome-wide association studies, the data contained within the many SNPs typed can be used to assess population stratification without the need to undertake any further genotyping. Several methods have been suggested to control for population stratification such as genomic control, structure association and EIGENSTRAT. These methods are discussed in Methodological quality of pharmacogenetic studies: issues of concern (Stat Med. 2008;27(30):6547-69), Softwares and methods for estimating genetic ancestry in human populations (Hum Genomics 2013;7(1):1) and Population Stratification in Genetic Association Studies (Curr Protoc Hum Genet. 2017;95:1.22.1–1.22.23).
The main advantage of exposure-only and case-only designs is the smaller sample size that is required, at the cost of not being able to study the main effects of drug exposure (case-only) or genetic variant (exposure-only) on the outcome. Furthermore, interaction can be assessed only on a multiplicative scale, whereas from a public health perspective, additive interactions are very relevant. Up till now GWAS with gene*interactions have not been very rewarding because of the required huge power. However, this is likely to improve as genetic data is linked to longitudinal clinical data in large biobanks, as described in Drug Response Pharmacogenetics for 200,000 UK Biobank Participants (Biocomputing 2021;184-95). An important condition that has to be fulfilled for case-only studies is that the exposure is independent of the genetic variant, e.g., prescribers are not aware of the genotype of a patient and do not take this into account, directly or indirectly (by observing clinical characteristics associated with the genetic variant). In the exposure-only design, the genetic variant should not be associated with the outcome, for example variants of genes coding for cytochrome p-450 enzymes. When these conditions are fulfilled and the main interest is in the drug-gene interaction, these designs may be an efficient option. In practice, case-control and case-only studies usually result in the same interaction effect as empirically assessed in Bias in the case-only design applied to studies of gene-environment and gene-gene interaction: a systematic review and meta-analysis (Int J Epidemiol. 2011;40(5):1329-41). The assumption of independence of genetic and exposure factors can be verified among controls before proceeding to the case-only analysis. Further development of the case-only design for assessing gene-environment interaction: evaluation of and adjustment for bias (Int J Epidemiol. 2004;33(5):1014-24) conducted sensitivity analyses to describe the circumstances in which controls can be used as proxy for the source population when evaluating gene-environment independence. The gene-environment association in controls will be a reasonably accurate reflection of that in the source population if baseline risk of disease is small (<1%) and the interaction and independent effects are moderate (e.g., risk ratio<2), or if the disease risk is low (e.g., <5%) in all strata of genotype and exposure. Furthermore, non-independence of gene-environment can be adjusted in multivariable models if non-independence can be measured in controls. Further methodological considerations and assumptions of study designs in pharmacogenomics research are discussed in A critical appraisal of pharmacogenetic inference (Clin Genet. 2018;93(3): 498-507).
Lastly, variation in prevalence and effect of pharmacogenetic variants across different ethnicities is an important consideration for study design and ultimately clinical utility, cost-effectiveness and implementation of testing. International research collaborations, as demonstrated in several studies (see HLA-B*5701 Screening for Hypersensitivity to Abacavir, N Engl J Med. 2008;358(6):568-79; and Effect of Genotype-Guided Oral P2Y12 Inhibitor Selection vs Conventional Clopidogrel Therapy on Ischemic Outcomes After Percutaneous Coronary Intervention: The TAILOR-PCI Randomized Clinical Trial, JAMA. 2020; 25;324(8):761-71), encourage greater representation of different populations and ensure broader applicability of pharmacogenomic study results. Diverse ethnic representation in study recruitment is important to detect the range of variant alleles of importance across different ethnic groups and reduce inequity in the clinical impact of pharmacogenomic testing once implemented.
16.3.4. Data collection
The same principles and approaches to data collection as for other pharmacoepidemiological studies can be followed (see Chapter 8 of this Guide on Approaches to Data Collection). An efficient approach to data collection for pharmacogenetic studies is to combine secondary use of electronic health records with primary data collection (e.g., collection of biological samples to extract DNA).
Examples are provided in SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink (Clin Pharmacol Ther. 2013;94(6):695-701), Diuretic therapy, the alpha-adducin gene variant, and the risk of myocardial infarction or stroke in persons with treated hypertension (JAMA. 2002;287(13):1680-9) and Interaction between the Gly460Trp alpha-adducin gene variant and diuretics on the risk of myocardial infarction (J Hypertens. 2009;27(1):61-8). Another approach to enrich electronic healthcare records with data from biological samples is record linkage to biobanks as illustrated in Genetic variation in the renin-angiotensin system modifies the beneficial effects of ACE inhibitors on the risk of diabetes mellitus among hypertensives (Hum Hypertens. 2008;22(11):774-80). A third approach is to use active surveillance methods to fully characterise drug effects such that a rigorous phenotype can be developed prior to genetic analysis. This approach was followed in Adverse drug reaction active surveillance: developing a national network in Canada's children's hospitals (Pharmacoepidemiol Drug Saf. 2009;18(8):713-21) and EUDRAGENE: European collaboration to establish a case-control DNA collection for studying the genetic basis of adverse drug reactions (Pharmacogenomics 2006;7(4):633-8).
16.3.5. Data analysis
The focus of data analysis should be on the measure of effect modification (see Chapter 7). Attention should be given to whether the mode of inheritance (e.g., dominant, recessive or additive) is defined a priori based on prior knowledge from functional studies. However, investigators are usually naïve regarding the underlying mode of inheritance. A solution might be to undertake several analyses, each under a different assumption, though the approach to analysing data raises the problem of multiple testing (see Methodological quality of pharmacogenetic studies: issues of concern, Stat Med. 2008;27(30):6547-69). The problem of multiple testing and the increased risk of type I error is in general a problem in pharmacogenetic studies evaluating multiple SNPs, multiple exposures and multiple interactions. The most common approach to correct for multiple testing is to use the Bonferroni correction. This correction may be considered too conservative and runs the risk of producing null results. Other approaches to adjust for multiple testing include permutation testing and false discovery rate (FDR) control, which are less conservative. The FDR, described in Statistical significance for genome-wide studies (Proc Natl Acad Sci. USA 2003;100(16):9440-5), estimates the expected proportion of false-positives among associations that are declared significant, which is expressed as a q-value.
Alternative innovative methods are becoming increasingly used, such as Mendelian Randomization (see Mendelian Randomization: New Applications in the Coming Age of Hypothesis-Free Causality, Annu Rev Genomics Hum Genet. 2015;16:327-50), systems biology, Bayesian approaches, data mining (see Methodological and statistical issues in pharmacogenomics, J Pharm Pharmacol. 2010;62(2):161-6) and polygenic risk scores (see Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations, Nat Genet. 2018;50(9):1219-1224; The potential of polygenic scores to improve cost and efficiency of clinical trials, Nat Commun. 2022;13(1):2922; Polygenic heterogeneity in antidepressant treatment and placebo response, Transl Psychiatry. 2022;12(1):456; Genetic risk and incident venous thromboembolism in middle-aged and older adults following COVID-19 vaccination, J Thromb Haemost. 2022;20(12):2887-2895).
Important complementary approaches include the conduct of individual patient data meta-analyses and/or replication studies to avoid the risk of false-positive findings.
An important step in analysis of genome-wide association studies data that needs to be considered is the conduct of rigorous quality control procedures before conducting the final association analyses. This becomes particularly important when phenotypic data were originally collected for a different purpose (“secondary use of data”). Relevant guidelines include Guideline for data analysis of genomewide association studies (Cancer Genomics Proteomics 2007;4(1):27-34) and Statistical Optimization of Pharmacogenomics Association Studies: Key Considerations from Study Design to Analysis (Curr Pharmacogenomics Person Med. 2011;9(1):41-66).
To improve both reproducibility, efficiency and interoperability across multiple data sources, the use of a common data model (CDM) is increasingly used in pharmacoepidemiological studies (see Chapter 9). Some healthcare databases with genomic data have been mapped to CDMs (see Transforming and evaluating the UK Biobank to the OMOP Common Data Model for COVID-19 research and beyond, J Am Med Inform Assoc. 2022;30(1):103-111). However, CDMs were developed for routinely collected healthcare and claims data, and thus, data on genetic variation are in general not yet integrated.
16.3.6. Reporting
The guideline STrengthening the Reporting Of Pharmacogenetic Studies: Development of the STROPS guideline (PLOS Medicine 2020;17(9):e1003344) should be followed for reporting findings of pharmacogenetic studies. Essential Characteristics of Pharmacogenomics Study Publications (Clin Pharmacol Ther. 2019;105(1):86-91) also provides recommendations to ensure that all the relevant information is reported in pharmacogenetic studies. As pharmacogenetic information is increasingly found in drug labels, as described in Pharmacogenomic information in drug labels: European Medicines Agency perspective (Pharmacogenomics J. 2015;15(3):201–10), it is essential to warrant consistency across the reporting of pharmacogenetic studies. Additional efforts by regulatory agencies, international organisations or boards to standardise the reporting and utilisation of pharmacogenetic studies will be discussed in the next section.
16.3.7. Clinical Implementation and Resources
An important step towards the implementation of the use of genotype information to guide pharmacotherapy is the development of clinical practice guidelines. A valuable pharmacogenomics knowledge resource is PharmGKB which curates and disseminates information about the impact of human genetic variation on drug responses, including genotype-phenotype relationships, potentially clinically actionable gene-drug associations, clinical guidelines, and drug labels. The development and publication of clinical practice guidelines for pharmacogenomics has been driven by international initiatives including the Clinical Pharmacogenetics Implementation Consortium, the European Medicines Agency Pharmacogenomics Working Party , and the DPWG: Dutch Pharmacogenetics Working Group. See also Pharmacogenetics: From Bench to Byte— An Update of Guidelines (Clin Pharmacol Ther. 2011;89(5):662–73); Use of Pharmacogenetic Drugs by the Dutch Population (Front Genet. 2019;10:567); and the Canadian Pharmacogenomics Network for Drug Safety. Evidence of clinical utility and cost-effectiveness of pharmacogenomic tests is important to support the translation of clinical guidelines into policies for implementation across health services, such as pharmacogenomic testing for DPYD polymorphisms with fluoropyrimidine therapies (see EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine).
The clinical implementation of pharmacogenomic testing requires consideration of complex clinical pathways and the multifactorial nature of drug response. Translational research and clinical utility studies can identify issues arising from the translation of pharmacokinetic or retrospective studies into real-world implementation of pharmacogenomic testing (see Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan, N Engl J Med. 2011;364(12):1126-33). Careful consideration is required in the interpretation of gene variants which cause a spectrum of effects. Binary interpretation or thresholds for phenotypic categorisation within clinical guidelines may result in different treatment recommendations for patients who would ultimately have the same drug response. In addition, the safety, efficacy and cost-effectiveness of alternative treatments are important factors in assessing the overall health benefit to patients from pharmacogenomic testing.
Further, some groups of patients may require specific treatment guidelines. Research studies such as the NICHD-funded Optimal Medication Management for Mothers with Depression (OPTI-MOM) aim to understand how best to manage drug therapy in pregnant women and investigate the impact of pharmacogenomics with the goal of generating treatment guidelines for proactive management during pregnancy (see Rationale and design for an investigation to optimize selective serotonin reuptake inhibitor treatment for pregnant women with depression, Clin Pharmacol Ther. 2016;100(1):31-3; and Pharmacogenomics in pregnancy. Semin Perinatol. 2020;44(3):151222).
Within clinical practice, the choice of technology for testing must be mapped to the clinical pathway to ensure that test results are available at an appropriate time to guide decision-making. Other key factors for clinical implementation include workforce education in pharmacogenomics, multidisciplinary pathway design, digital integration and tools to aid shared decision making (see Attitudes of clinicians following large-scale pharmacogenomics implementation, Pharmacogenomics J. 2016;16(4):393-8; Pharmacogenomics Implementation at the National Institutes of Health Clinical Center, J Clin Pharmacol. 2017;57 (Suppl 10):S67-S77; The implementation of pharmacogenomics into UK general practice: a qualitative study exploring barriers, challenges and opportunities, J Community Genet. 2020;11(3):269-77; Implementation of a multidisciplinary pharmacogenomics clinic in a community health system, Am J Health Syst Pharm. 2016;73(23):1956-66).
Large-scale international population studies of clinical utility in pharmacogenomics will contribute to understanding these real-world implementation factors, including studies underway with the U-PGx (see Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium, Clin Pharmacol Ther. 2017;101(3):341-58) and The IGNITE Pharmacogenetics Working Group: An Opportunity for Building Evidence with Pharmacogenetic Implementation in a Real-World Setting, Clin Transl Sci. 2017;10(3):143-6).
The clinical utility of pharmacogenetic testing before starting drug treatment is well documented for several single gene–drug pairs. To further improve the understanding of how genetic variation may increase the risk of adverse drug reactions, the Medicines and Healthcare Products Regulatory Agency (MHRA) together with Genomics England launched the Yellow Card biobank, which will contain genetic data and patient samples, and will operate alongside the MHRA’s Yellow Card reporting site for suspected side effects and adverse incidents involving medicines and medical devices.
Beyond single gene genotyping, a recent study has investigated the clinical benefit of using a pharmacogenetic panel to guide prescription and showed that pharmacogenetics-guided prescribing resulted in a 30% reduction of clinically relevant adverse drug reactions (see A 12-gene pharmacogenetic panel to prevent adverse drug reactions: an open-label, multicentre, controlled, cluster-randomised crossover implementation study, Lancet. 2023;401(10374):347-356). Lastly, international networks on pharmacogenomics research provide biological insights into emerging diseases and can support public health actions. For example, the COVID-19 Host Genetics Initiative (The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic, Eur J Hum Genet. 2020; 28(6): 715–8) has enabled rapid genetic association studies on COVID-19 and advanced the global knowledge of SARS-CoV-2 infection by creating a common repository for COVID-19 genetic studies (https://www.covid19hg.org/) and performing powered meta-analyses (Mapping the human genetic architecture of COVID-19, Nature 2021; 600:472-7). Although the discovery of genetic variants associated with susceptibility and severity of COVID-19 disease is challenged by the accurate ascertainment of cases and controls (Understanding COVID-19 through genome-wide association studies, Nature Genetics 2022; 54:368–9), the COVID-19 HGI identified novel host genetic factors associated with COVID-19 and created a framework for international collaboration for future genetic discoveries in emerging pandemics.
16.4. Methods for pharmacovigilance impact research
Note: Chapter 16.4. (formerly 15.4.) has not been updated for Revision 11 of the Guide, as contents remain up-to-date.
16.4.1. Introduction
Pharmacovigilance activities aim to protect patients and promote public health. This includes implementing risk minimisation measures that lead to changes in the knowledge and behaviour of individuals (e.g. patients, consumers, caregivers and healthcare professionals) and in healthcare practice. Impact research aims to generate evidence to evaluate the outcomes of these activities which may be intended or unintended. This approach has been adopted in the EMA Guideline on good pharmacovigilance practices (GVP) - Module XVI – Risk minimisation measures: selection of tools and effectiveness indicators (Rev 2), which is currently undergoing revision (see Guideline on good pharmacovigilance practices (GVP) - Module Risk Minimisation Measures for the draft of Rev. 3).
Pharmacovigilance activities are frequently examined for their impact on processes of healthcare delivery, such as healthcare outcomes or drug utilisation patterns following changes to the product information. In addition, measuring dissemination of risk minimisation is of importance as well as changes in knowledge, awareness and behaviour of healthcare professionals and patients.
These effects can be assessed separately, or combined in a framework, which is more challenging and therefore rarely done. An example of such a standardised framework includes evaluation of the effectiveness of risk minimisation measures through four domains: data, knowledge, behaviour and outcomes (Evaluating the effectiveness of risk minimisation measures: the application of a conceptual framework to Danish real-world dabigatran data; Pharmacoepidemiol Drug Saf. 2017;26(6):607-14). Further testing of this method is needed, however, to ascertain its usefulness in regulatory practice.
Measuring the impact of pharmacovigilance activities may be challenging as these activities may target stakeholder groups at different levels of the healthcare system, co-exist with other unrelated events that can influence healthcare, and can use several tools applied simultaneously or sequentially to deliver information and influence behaviour (Measuring the impact of pharmacovigilance activities, challenging but important; Br J Clin Pharmacol. 2019;85(10):2235-7). In addition to the intended outcomes of pharmacovigilance activities, there may be unintended outcomes which are important to be measured as they could counteract the effectiveness of risk minimisation. Another challenging aspect is separating the outcomes of individual pharmacovigilance activities from simultaneous events such as media attention, reimbursement policies, publications in scientific journals, changes in clinical guidelines and practice, or secular trends in health outcomes.
This Chapter provides a detailed guidance on the methodological conduct of impact studies.
16.4.2. Outcomes
Outcomes to be studied in impact research are closely tied to the nature and objective of the pharmacovigilance activities. Because regulatory actions are mostly tailored to individual medicinal products, there is no standard outcome that could be measured for each activity and the concepts outlined in this chapter need to be applied on a case-by-case basis (Post-approval evaluation of effectiveness of risk minimisation: methods, challenges and interpretation; Drug Saf. 2014;37(1):33-42).
Outcome measures provide an overall indication of the level of risk reduction that has been achieved with a specific risk minimisation measure in place. This may also require measuring outcomes not linked to the specific medicinal product but representing potential unintended consequences of regulatory interventions e.g., change of non-target drug use in a population leading to less favourable health outcomes. Examples are provided in Table XVI.1 of the Guideline on good pharmacovigilance practices (GVP) - Module Risk Minimisation Measures.
Relevant outcomes may include: information dissemination and risk knowledge; changes in behaviour or clinical practice; drug utilisation patterns (e.g. prescribing or dispensing rates, use of treatment alternatives); and health outcomes (Measuring the impact of medicines regulatory interventions - Systematic review and methodological considerations; Br J Clin Pharmacol. 2018;84(3):419-33).
Dissemination of information and risk knowledge can be assessed in a quantitative, qualitative or mixed-methods manner. Quantitative assessment can involve measuring the proportion of healthcare professionals and patients aware of the risk minimisation measure as well as their level of comprehension (Effectiveness of Risk Minimization Measures to Prevent Pregnancy Exposure to Mycophenolate-Containing Medicines in Europe; Pharmaceut Med. 2019;33(5):395-406). Qualitative measures often focus on understanding of attitudes about the risk minimisation measure, impact of external factors on implementation and information update whilst mixed methods utilise both qualitative and quantitative approaches.
Assessment of behavioural changes is performed to measure if changes towards intended behaviour have been achieved, and to what extent. These measures align with those applied when measuring dissemination of information and risk knowledge. Quantitative assessment can include measuring the proportion of patients exposed to a medicinal product which is not in accordance with authorised use (off label use, contraindicated use, interactions). A qualitative assessment may allow an in-depth understanding of enablers and barriers in relation to awareness, attitudes towards use of the medicinal product and the causes why intended outcomes may not have been achieved.
Health outcomes should preferably be measured directly. They may include clinical outcomes such as all-cause mortality, congenital defects or other conditions that prompted the pharmacovigilance activity. Direct measurement of health outcomes is not always feasible or may not be necessary, for example when it can be replaced with indirect measures. Indirect surrogate measures may use data on hospitalisations, emergency department admissions or laboratory values e.g. blood pressure as a surrogate for cardiac risk, as outlined in Practical Approaches to Risk Minimisation for Medicinal Products: Report of CIOMS Working Group IX. An example of use of a surrogate measure is glycaemic outcomes (HbA1C change from baseline) in patients with diabetes mellitus using the Veterans Integrated Services Network database; the results confirmed a 45% discontinuation of thiazolidinedione use in this population and a worsening of glycaemic control following safety warning publicity in 2007, which may have driven the decline in usage of this class of medicines (Impact of thiazolidinedione safety warnings on medication use patterns and glycemic control among veterans with diabetes mellitus; J Diabetes Complications 2011;25(3):143-50).
Depending on the nature of the safety concern and the regulatory intervention, or when the assessment of patient-relevant health outcomes is unfeasible (e.g. inadequate number of exposed patients, rare adverse reaction), the dissemination of safety information, risk knowledge or behavioural changes may be alternative objectives of impact research (Guideline on good pharmacovigilance practices (GVP) - Module VIII – Post-authorisation safety studies (Rev 3).
16.4.3. Considerations on data sources
The impact of pharmacovigilance activities can be measured using both primary and secondary data collection, although the literature shows that the latter is more commonly used (Measuring the impact of medicines regulatory interventions - Systematic review and methodological considerations; Br J Clin Pharmacol. 2018;84(3):419-33). Chapter 7 of this Guide provides a general description of the main characteristics, advantages and disadvantages of various data sources. Chapter 7.1.2. provides guidance on primary data collection through surveys.
The impact of pharmacovigilance activities should be interpreted with a view to the limitations of the data sources used for the evaluation (A General Framework for Considering Selection Bias in EHR-Based Studies: What Data Are Observed and Why?; EGEMS. (Wash DC.) 2016;4(1):1203). Researchers should have a clear understanding of the limitations of the different data sources when planning their research and assess whether these limitations could impact the results in one direction or the other in such a way that their interpretation may be significantly influenced, for example due to bias or unmeasured confounders. As for all observational studies, the evaluation of the usefulness and limitation of a given data source for the study requires a very good understanding of the research question.
Primary data collection, via interviews or surveys, can usually never cover the complete target population. Therefore, a sampling approach is often required which can involve those that prescribe, dispense or use the medicinal product. Sampling should be performed in accordance with the Guideline on good pharmacovigilance practices (GVP) - Module XVI Addendum II, ensuring target population representativeness. The following elements should be considered to minimise bias and optimise generalisability: sampling procedures (including sample size), design and administration of the data collection instrument, analytical approaches and overall feasibility (including ethics).
Different databases are unlikely to capture all impact–relevant outcomes, even when they are linked to one another. Data of good quality may be available on hard outcomes such as death, hospital admission, emergency room visit or medical contacts but claims databases rarely capture primary care diagnoses, symptoms, conditions or other events that do not lead to a claim, such as suicidal ideation, abuse or misuse. An accurate definition of the outcomes also often requires the development of algorithms that need validation in the database that will be used for impact measurement.
Nurse-Led Medicines' Monitoring for Patients with Dementia in Care Homes: A Pragmatic Cohort Stepped Wedge Cluster Randomised Trial (PLoS One 2015;10(10):e0140203) reported that only about 50% of the less serious drug-related problems listed in the product information are recorded in patient notes. If generalisable to electronic data sources, this would indicate that incomplete recording of patient-reported outcomes of low severity may reduce the likelihood of identifying some outcomes related to a pharmacovigilance activity, for example a change in the frequency of occurrence of an adverse drug reaction (ADR). Combining different approaches such as integrating a patient survey would be necessary to overcome this situation.
Missing information on vulnerable populations, such as pregnant women, and missing mother-child or father-child links is a significant barrier to measuring the impact of paternal/maternal exposure or behaviour. For example, the impact of pregnancy prevention programmes could not be accurately assessed using European databases that had been used to report prescribing in pregnancy (The limitations of some European healthcare databases for monitoring the effectiveness of pregnancy prevention programmes as risk minimisation measures; Eur J Clin Pharmacol. 2018;74(4):513-20). This was largely due to inadequate data on planned abortions and exposure to oral contraceptives.
Depending on the initial purpose of the data source used for impact research, information on potential confounders may be missing, such as indication of drug use, co-morbidities, co-medication, smoking, diet, body mass index, family history of disease or recreational drug use. Missing information may impair a valid assessment of risk factors for changes in health care practice, but this limitation should be considered in light of the research question. In some settings, record linkage between different types of data sources including different information could provide comprehensive data on the frequency of ADRs and potential confounders (Health services research and data linkages: issues, methods, and directions for the future; Health Serv Res. 2010;45(5 Pt 2):1468-88; Selective Serotonin Reuptake Inhibitor (SSRI) Antidepressants in Pregnancy and Congenital Anomalies: Analysis of Linked Databases in Wales, Norway and Funen, Denmark; PLoS One 2016;11(12):e0165122; Linking electronic health records to better understand breast cancer patient pathways within and between two health systems; EGEMS. (Wash DC.) 2015;3(1):1127).
16.4.4. Study designs
16.4.4.1. Single time point cross-sectional study
The cross-sectional study design as defined in Appendix 1.1.2.1 of the Guideline on good pharmacovigilance practices (GVP) - Module VIII – Post-authorisation safety studies (Rev 3) collects data at a single point in time after implementation of a regulatory intervention. However, cross-sectional studies have limitations as a sole measure of the impact of interventions. Cross-sectional studies may include data collected through surveys and can be complemented with data from other studies, e.g. on patterns of drug use (Healthcare professional surveys to investigate the implementation of the isotretinoin Pregnancy Prevention Programme: a descriptive study; Expert Opin Drug Saf. 2013;12(1):29-38; Prescriptive contraceptive use among isotretinoin users in the Netherlands in comparison with non-users: a drug utilisation study; Pharmacoepidemiol Drug Saf. 2012;21(10):1060-6).
16.4.4.2. Before-and-after study
A before-and-after study is defined as an evaluation (at one point in time) before and (one point in time) after the date of the intervention and/or its implementation. When uncontrolled, before-and-after studies need to be interpreted with caution as any baseline trends are ignored, potentially leading to the intervention effect being incorrectly estimated. Including a control (e.g., a population that did not receive the intervention or a drug not targeted by the risk minimisation measure) can strengthen this design by minimising potential confounding. However, identifying a suitable control group may be challenging or unfeasible as any regulatory action aimed at reducing risk is intended to be applied to the entire target population (see Post-approval evaluation of effectiveness of risk minimisation: methods, challenges and interpretation; Drug Saf. 2014;37(1):33-42 and Measuring the impact of medicines regulatory interventions - Systematic review and methodological considerations; Br J Clin Pharmacol. 2018;84(3):419-33). When a suitable control group is available, the difference-in-differences (DiD) method can be used. The DiD method is a controlled before-and-after design whereby comparisons are made between two similar groups under different conditions. The outcome can be measured either at a single pre-intervention and post-intervention time point, or by comparing pre- and post-intervention means, but it does not incorporate time. The DiD method then takes the difference for both groups (exposed and control) before and after the intervention, thereby controlling for varying factors in estimating the impact of the intervention (see The use of controls in interrupted time series studies of public health interventions; Int J Epidemiol 2018;47:2082–93 and Difference-in-Differences Method in Comparative Effectiveness Research: Utility with Unbalanced Groups; Appl Health Econ Health Policy. 2016; 14: 419–29). The DiD method relies upon the assumption that both groups are similar and trends are parallel, hence may be susceptible to residual confounding as a result of differences between the groups.
16.4.4.3. Time series design
A time series is a sequence of data points (values) usually gathered at regularly spaced intervals over time. These data points can represent a value or a quantification of outcomes that are used for impact research. The underlying trend of a particular outcome is ‘interrupted’ by a regulatory intervention at a known point in time. Time series data can be analysed using various methods, including interrupted time series (ITS) and Joinpoint analysis.
16.4.4.4. Cohort study
The cohort study design as defined in Appendix 1.1.2.2 of the Guideline on good pharmacovigilance practices (GVP) - Module VIII – Post-authorisation safety studies (Rev 3) can be useful in impact research to establish the base population for the conduct of drug utilisation studies or to perform aetiological studies.
Cohort studies can be used to study exposure to the medicine targeted by regulatory interventions before and after its implementation, and indeed to perform drug utilisation studies in clinical populations targeted by these interventions. To model their impact on health outcomes, more complex study designs may be required, that are the subject of further research.
The following are examples of cohort studies being used for:
-
Impact research evaluating pregnancy prevention programmes (Isotretinoin exposure during pregnancy: a population-based study in The Netherlands; BMJ. Open 2014;4(11):e005602);
-
Drug utilisation in target populations (Impact of EMA regulatory label changes on systemic diclofenac initiation, discontinuation, and switching to other pain medicines in Scotland, England, Denmark, and The Netherlands; Drug Saf. 2020;29(3):296-305);
-
Aetiological studies examining the impact on health outcomes (Measuring the Effectiveness of Safety Warnings on the Risk of Stroke in Older Antipsychotic Users: A Nationwide Cohort Study in Two Large Electronic Medical Records Databases in the United Kingdom and Italy; Drug Saf. 2019;42(12):1471-85).
16.4.4.5. Randomised controlled trial
The randomised controlled trial (RCT) as defined in Appendix 1.1.2.2 of the Guideline on good pharmacovigilance practices (GVP) - Module VIII – Post-authorisation safety studies (Rev 3) can be useful in evaluating the effectiveness of different interventions but it is not always possible to randomise individual participants and few examples exist (Improved therapeutic monitoring with several interventions: a randomized trial; Arch Intern Med. 2006;166(17):1848-54). Designs including cluster randomised trials or step-wedge trials may be more feasible, in which randomisation is conducted at the level of organisation, when a phased roll-out is being considered (Research designs for studies evaluating the effectiveness of change and improvement strategies; Qual Saf Health Care 2003;12(1):47-52). RCTs could be considered more often to generate evidence on the impact of pharmacovigilance interventions by evaluating interventions that potentially enhance agreed safety information and normal methods of dissemination and communication channels.
16.4.5. Analytical methods
The analytical methods to be applied in impact research depend on the study design and approach to data collection. Various types of analyses have been used to assess the impact of a regulatory guidance, as described in: Measuring the impact of medicines regulatory interventions - Systematic review and methodological considerations (Br J Clin Pharmacol. 2018;84(3):419-33); Impact of regulatory guidances and drug regulation on risk minimization interventions in drug safety: a systematic review (Drug Saf. 2012;35(7):535-46); and A descriptive review of additional risk minimisation measures applied to EU centrally authorised medicines 2006-2015 (Expert Opin Drug Saf. 2017;16(8):877-84).
16.4.5.1 Descriptive statistics
Descriptive measures are the basis of quantitative analyses in studies evaluating the impact of regulatory interventions. Whilst appropriate to describe the population to understand generalisability, simple descriptive approaches do not determine whether statistically significant changes have occurred (Measuring the impact of medicines regulatory interventions - Systematic review and methodological considerations; Br J Clin Pharmacol. 2018;84(3):419-33). When simple descriptive statistics are used, they are often insufficiently valid to determine statistical significance.
16.4.5.2 Time series analysis
Interrupted time series (ITS) analysis
ITS analysis, sometimes referred to as interrupted segmented regression analysis, can provide statistical evidence about whether observed changes in a time series represent a real decrease or increase by accounting for secular trends. ITS has commonly been used to measure the impact of regulatory interventions and is among the more robust approaches to pharmacovigilance impact research (Measuring the impact of medicines regulatory interventions - Systematic review and methodological considerations; Br J Clin Pharmacol. 2018;84(3):419-33; Impact of EMA regulatory label changes on systemic diclofenac initiation, discontinuation, and switching to other pain medicines in Scotland, England, Denmark, and The Netherlands; Pharmacoepidemiol Drug Saf. 2020;29(3):296-305; The Effect of Safety Warnings on Antipsychotic Drug Prescribing in Elderly Persons with Dementia in the United Kingdom and Italy: A Population-Based Study; CNS Drugs 2016;30(11):1097-109).
ITS is well suited to study changes in outcomes that are expected to occur relatively quickly following an intervention, such as change in prescribing, and can consist of averages, proportions, counts or rates. ITS can be used to estimate a variety of outcomes including: the immediate change in outcome after the intervention; the change in trend in the outcome compared to before the intervention; and the effects at specific time periods following the intervention.
Common segmented regression models fit a least squares regression line to each time segment and assume a linear relationship between time and the outcome within each segment.
When the effects of interventions take time to manifest, this can be accounted for through the use of lag times in the analysis to avoid incorrect specification of the intervention effect. To model these effects, one can exclude from the analysis outcome values that occur during the lag or during the intervention period. Alternatively, with enough data points, the period may be modelled as a separate segment.
ITS regression requires that the time point of the intervention is known prior to the analysis and sufficient data points are collected before and after the intervention for adequate power. Studies with a small number of data points should be interpreted with caution as they may be underpowered.
An assumption of ITS segmented regression analysis is that time points are independent of each other. Autocorrelation is a measure of how correlated data collected closely together in time are with each other. If autocorrelation is present, it may violate the underlying model assumptions that observations are independent of each other and can lead to an over-estimation of the statistical significance of effects. Autocorrelation can be checked by examining autocorrelation and partial autocorrelation function plots and checking the Durbin-Watson statistic or performing the Breusch-Godfrey test (Testing for serial correlation in least squares regression. I; Biometrika. 1950;37(3-4):409-28; Testing for serial correlation in least squares regression. II; Biometrika. 1951;38(1-2):159-78). Factors such as autocorrelation, seasonality and non-stationarity should therefore be checked and may require more complicated modelling approaches if detected, e.g. autoregressive integrated moving average (ARIMA) models (Impact of FDA Black Box Warning on Psychotropic Drug Use in Noninstitutionalized Elderly Patients Diagnosed With Dementia: A Retrospective Study; J Pharm Pract. 2016;29(5):495-502; IMI Work Package 5: Benefit –Risk Integration and Visual Representation).
Long time periods may also be affected by historical changes in trend that can violate model assumptions. Therefore, data should always be visually inspected and reported.
Data point outliers that are explainable, such a sudden peak in drug dispensing in anticipation of a drug restriction policy can be controlled for using an indicator term. Outliers that result from random variation can be treated as regular data point.
Another caveat when conducting ITS analysis relates to possible outcome measure ceiling or floor effects. For example, when studying the impact of an intervention in improving the proportion of patients treated with a drug, the outcome has a natural ceiling of 100% and thus, depending of the initial level of measurement, minimal change in the outcome is observed.
Time-varying confounding, such as from concomitant interventions, may be addressed by use of a control outcome in the same population or a control population using the same outcome. An advantage on ITS analysis is the ease in stratifying results by different groups.
Joinpoint analysis
Accurately establishing the date of the intervention time period may be challenging (e.g. during a phased roll out of a regulatory intervention or when attempting to assess different parts of a regulatory intervention). In such instances, more complex modelling techniques and other approaches time series approaches could be considered.
Statistical analysis using joinpoint regression identifies the time point(s) where there is a marked change in trend (the ‘joinpoint’) in the time series data and estimates the regression function compared with previously identified joinpoints. Joinpoints can be identified by using permutation tests using Monte Carlo methods or Bayesian Information Criterion approaches (Permutation tests for joinpoint regression with applications to cancer rates; Stat Med. 2000;19(3):335-51). As the final number of joinpoints is established on the basis of a statistical criterion, their position is not fixed. Therefore, joinpoint regression does not require that the date of the regulatory intervention is pre-specified. It can be used to estimate the average percent change in an outcome, which is a summary measure of the trend over a pre-specified fixed interval. It can also be used to undertake single or pairwise comparisons.
16.4.5.3 Other statistical techniques
Different types of regression models can be applied to the time series data once it has been properly organised depending upon the exact question being asked such as Poisson regression (Interrupted time series regression for the evaluation of public health interventions: a tutorial; Int J Epidemiol. 2017;46(1):348-55. Erratum in: Int J Epidemiol. 2020;49(4):1414). These methods are based on the assumption that error terms are normally distributed. When time series analysis measurements are based at extreme values (e.g. all are near 0% or near 100% or with low cell counts near 0) alternative approaches may be required (e.g. aggregate binomial regression models) and advice from an experienced statistician is recommended.
16.4.5.4 Examples of impact research using time series analysis
Before-and-after after time series have been used to evaluate the effects of:
-
Paracetamol pack size reductions introduced in the UK in 1998 on poisoning deaths and liver transplants (Long term effect of reduced pack sizes of paracetamol on poisoning deaths and liver transplant activity in England and Wales: interrupted time series analyses; BMJ. 2013;346:f403);
-
Black Triangle Label on Prescribing of New Drugs in the United Kingdom (Impact of the black triangle label on prescribing of new drugs in the United Kingdom: lessons for the United States at a time of deregulation; Pharmacoepidemiol Drug Saf. 2017;26(11):1307-13);
-
FDA boxed warning on the duration of use for depot medroxprogesterone acetate (The impact of the boxed warning on the duration of use for depot medroxprogesterone acetate; Pharmacoepidemiol Drug Saf. 2017;26(7):827-36);
-
Withdrawal of fusafungine from the market on prescribing of antibiotics, other nasal or throat preparations and tyrothricin in Germany (Effect of withdrawal of fusafungine from the market on prescribing of antibiotics and other alternative treatments in Germany: a pharmacovigilance impact study; Eur J Clin Pharmacol. 2019;75(7):979-84);
-
FDA black box warning on fluoroquinolone and alternative antibiotic use in southeastern US hospitals (Impact of FDA black box warning on fluoroquinolone and alternative antibiotic use in southeastern US hospitals; Infect Control Hosp Epidemiol. 2019;40(11):1297-1300);
-
A re-analysis of published UK impact studies showed that UK regulatory risk communications were associated with significant changes in targeted prescribing and potential changes in clinical outcomes (Impact of medicines regulatory risk communications in the UK on prescribing and clinical outcomes: Systematic review, time series analysis and meta-analysis; Br J Clin Pharmacol. 2020;86(4):698-710).
Examples of the use of Joinpoint regression analysis:
-
Scientific publications, FDA advisories and media exposure on glitazone use (Changes in glitazone use among office-based physicians in the U.S., 2003-2009; Diabetes Care. 2010;33(4):823-5);
-
The fall of hormone replacement therapy in England following the results of the women’s health initiative (What was the immediate impact on population health of the recent fall in hormone replacement therapy prescribing in England? Ecological study; J Public Health (Oxf.). 2010;32(4):555-64).
16.4.5.5 Regression modelling
Multivariable regression allows controlling for potential confounding factors or to study factors associated with the impact or non-impact of regulatory interventions.
An analysis with multivariate regression was used in Measuring the Effectiveness of Safety Warnings on the Risk of Stroke in Older Antipsychotic Users: A Nationwide Cohort Study in Two Large Electronic Medical Records Databases in the United Kingdom and Italy (Drug Saf. 2019;42(12):1471-85). The Medicines and Healthcare Regulatory Agency (MHRA) and the Italian Drug Agency (AIFA) both launched a safety warning on the risk of stroke and all-cause mortality with antipsychotics in older people with dementia. In the UK, the MHRA launched a warning in March 2004 for the use of risperidone and olanzapine which was expanded to all antipsychotics in March 2009. In Italy, AIFA restricted prescribing of antipsychotics in the elderly to specific prescribing centres in July 2005, which was followed by communication about these restrictions in May 2009. A retrospective new-user cohort study was undertaken to estimate incidence rates of stroke in elderly incident antipsychotic users. The authors showed a significant reduction of stroke after both safety warnings in the UK, while there was no impact of the warning on incidence rates of stroke in Italy. Metabolic screening in children receiving antipsychotic drug treatment (Arch Pediatr Adolesc Med. 2010;164(4):344-51) measured the impact of a class warning issued by the Food and Drug Administration (FDA) for all second-generation antipsychotics (SGAs) regarding the risk of hyperglycaemia and diabetes mellitus in 2003. This warning stated that glucose levels should be monitored in at-risk patients. A retrospective new-user cohort study was undertaken to estimate population-based rates of glucose and lipid testing in children after the availability of FDA warnings and to identify predictors of the likelihood of receiving glucose or lipid testing among SGAs-treated children after adjusting for covariates. Children without diabetes taking albuterol but no SGA drugs were used as controls. The authors showed that most included children starting treatment with SGAs did not receive recommended glucose and lipid screening.
More sophisticated methodologies, such as propensity-score matching (Chapter 5.2.3.2), instrumental variable analysis (Chapter 5.2.3.3) and time-varying exposures and covariates (Chapter 5.2.3.5) may be implemented in regression analyses if relevant.
Whichever design and method of analysis is used, consideration should be given to reporting both relative and absolute effects.
16.4.5.6 Other types of analytical methods
Metrics such as “Population Impact Number of Eliminating a Risk factor over time t” (PIN-ER-t), and “Number of Events Prevented in a Population” (NEPP) have proven valuable in assessing the impact of removing a risk factor on public health, and may be useful in assessing impact of regulatory interventions. Illustrative examples for population impact analyses include Potential population impact of changes in heroin treatment and smoking prevalence rates: using Population Impact Measures (Eur J Public Health 2009;19(1):28-31) and Assessing the population impact of low rates of vitamin D supplementation on type 1 diabetes using a new statistical method (JRSM Open 2016;7(11):2054270416653522). Further, statistical analysis using impact metrics is possible where proxy measures are used to assess the impact that one event or resource has on another, as shown in Communicating risks at the population level: application of population impact numbers (BMJ. 2003;327(7424):1162-5); the benefit-risk case study report for rimonabant in IMI Work Package 5: Benefit –Risk Integration and Visual Representation; and in Population Impact Analysis: a framework for assessing the population impact of a risk or intervention (J Public Health (Oxf.) 2012;34(1):83-9).
Predictive modelling techniques may provide an insight into future impact of regulatory actions. Modelling the risk of adverse reactions leading to product withdrawal alongside drug utilisation data can assess the number of patients at risk of experiencing the adverse reactions per year, and provide an estimate of the number of patients per year which are protected from as a result of regulatory action (Population Impact Analysis: a framework for assessing the population impact of a risk or intervention; J Public Health (Oxf.) 2012;34(1):83-9; Assessing the population impact of low rates of vitamin D supplementation on type 1 diabetes using a new statistical method; JRSM Open 2016;7(11):2054270416653522).
Chronographs, typically used for rapid signal detection in observational longitudinal databases, have been used to visualise the impact of regulatory actions. Although this is a novel method that could potentially be applied to rapidly assess impact, the method lacks ways to control for confounding. In addition, further validation may be required to understand in which situations this works well or not (A Novel Approach to Visualize Risk Minimization Effectiveness: Peeping at the 2012 UK Proton Pump Inhibitor Label Change Using a Rapid Cycle Analysis Tool; Drug Saf. 2019;42(11):1365-76).
16.4.6. Measuring unintended effects of regulatory interventions
Pharmacovigilance activities can have unintended consequences, which could in some cases counteract the effectiveness of risk minimisation measures. To determine the net attributable impact of pharmacovigilance activities, besides the intended outcomes, other outcomes associated with potential unintended consequences may need to be measured and incorporated into the design of impact research (see Table XVI.1 of the Guideline on good pharmacovigilance practices (GVP) - Module Risk Minimisation Measures). Examples of such studies include the Effect of withdrawal of fusafungine from the market on prescribing of antibiotics and other alternative treatments in Germany: a pharmacovigilance impact study (Eur J Clin Pharmacol. 2019;75(7):979-84), which was associated with an increase in prescribing of other nasal or throat preparations but no increase in alternative antibiotic prescribing. Another example concerns the unintended increased use of conventional antipsychotics in two European countries after the introduction of EU risk minimisation measures for the risk of stroke and all-cause mortality with atypical antipsychotic drug use (The Effect of Safety Warnings on Antipsychotic Drug Prescribing in Elderly Persons with Dementia in the United Kingdom and Italy: A Population-Based Study; CNS Drugs 2016;30(11):1097-109). Further, prescribers may extrapolate warnings for one group of patients to other groups (spill-over effects), although they may not share the same risk factors. In 2003, the FDA warned of an association between SSRI prescription and suicidality in paediatric patients (<18 years of age). Subsequently, the number of prescriptions of SSRIs in newly diagnosed adult patients fell without compensation by alternative medicines or treatment (Spillover effects on treatment of adult depression in primary care after FDA advisory on risk of pediatric suicidality with SSRIs; Am J Psychiatry 2007;164(8):1198-205).
Socio-economic factors may also play an important role in implementing regulatory interventions at local level. It has been suggested that practices in affluent communities are more likely to implement regulatory interventions faster than over-stretched or under-resourced practices in more deprived communities and that permanent changes in daily practice in these communities may take longer (THE INTERNATIONAL MARCÉ SOCIETY FOR PERINATAL MENTAL HEALTH BIENNIAL SCIENTIFIC CONFERENCE; Arch Womens Ment Health 2015;18:269–408; Prescribing of antipsychotics in UK primary care: a cohort study; BMJ Open 2014;4(12):e006135).
Both health care service providers and users may circumvent or ‘work round’ restrictions. Where medicines are restricted or restrictions are perceived as inconvenient, patients may turn to buying medicines over the internet, self-medicating with over-the-counter medicines or using herbals or other complementary medicines. Healthcare professionals may subvert requirements for additional documentation by realigning diagnostic categories (Changes in rates of recorded depression in English primary care 2003-2013: Time trend analyses of effects of the economic recession, and the GP contract quality outcomes framework (QOF); J Affect Disord. 2015;180:68-78) or switch to medicines where patient monitoring is not mandated (Incorporating Comprehensive Management of Direct Oral Anticoagulants into Anticoagulation Clinics; Pharmacotherapy 2017;37(10):1284-97). The effects of progressive dextropropoxyphene withdrawal in the EU since 2007 on prescribing behaviour showed an increased use of same level analgesics but also an increased use of paracetamol as monotherapy. Aggregated dispensation data suggested that the choice of analgesics depended on physician speciality, healthcare setting, indication, patients’ comorbidities and age, underlining the complexity and international differences in pain management (Use of analgesics in France, following dextropropoxyphene withdrawal; BMC Health Serv Res. 2018;18(1):231).
16.5. Artificial intelligence in pharmacoepidemiology
16.5.1. Introduction
Artificial intelligence (AI) is a catch-all term for a set of tools and techniques that allow machines to do activities commonly described as requiring human-level intelligence. While no consensus on a definition of AI exists, a common trend is an analogy to human intelligence, however, this is unhelpful as it suggests an idea of Artificial General Intelligence, whereas current techniques and tools are dedicated to assist specific tasks, i.e., Artificial Narrow Intelligence.
Machine Learning (ML) is considered a subset of AI and reflects the ability of computers to identify and extract rules from data rather than those rules being explicitly coded by a human. Deep Learning (DL) is a subtype of ML with increased complexity of how it parses and analyses data. The rules identified by ML or DL applications constitute an algorithm and the outputs are often said to be data-driven, as opposed to rules explicitly coded by a human that form knowledge-based algorithms.
Natural language processing (NLP) sits at the interface of linguistics, computer science and AI and is concerned with providing machines with the ability to understand text and spoken words. NLP can be subset into statistical NLP, which uses ML or DL approaches and symbolic NLP, which uses a semantic rule-based methodology. Applications of AI in pharmacoepidemiology can be broadly classified into those that extract and structure some data and those that produce some insight.
16.5.2. Applications of AI in pharmacoepidemiology
16.5.2.1. Data extraction
AI techniques can be used to extract text data from unstructured documents transforming it into information available in a structured, research-ready format to which statistical techniques can be applied. A potential application being explored is in extracting data from medical notes, usually including a named-entity recognition, i.e., discovering mentions of entities of a specific class or group such as medication or diseases, and a relation extraction, allowing to relate sets of entities, e.g., a medicine and an indication.
The 2010 i2b2/VA challenge on concepts, assertions, and relations in clinical text (J Am Med Inform Assoc. 2011;18(5):552-6) presents three tasks: a concept extraction of medical concepts from patient reports; a classification task focused on assigning assertion types for medical problem concepts; and a relation classification task focused on assigning relation types that hold between medical problems, tests, and treatments. Multiple algorithms were compared showing promising results for concept extraction. In NEAR: Named entity and attribute recognition of clinical concepts (J Biomed Inform. 2022;130:104092), three DL models were created for the same data used in the 2010 i2b2 challenge and have showed an improvement in performance.
Some of the first applications of ML and NLP to extract information from clinical notes focused on the identification of adverse drug events in medical notes, as illustrated in publications such as A method for systematic discovery of adverse drug events from clinical notes (J Am Med Inform Assoc. 2015;22(6):1196-204), Detecting Adverse Drug Events with Rapidly Trained Classification Models (Drug Saf. 2019;42(1):147-56) and MADEx: A System for Detecting Medications, Adverse Drug Events, and Their Relations from Clinical Notes (Drug Saf. 2019;42(1):123-33).
Another common application for medical concept extraction from clinical text is the identification of a relevant set of patients, often referred to as computable phenotyping as exemplified in Mayo clinical Text Analysis and Knowledge Extraction System (cTAKES): architecture, component evaluation and applications (J Am Med Inform Assoc. 2010;17(5):507-13). Combining deep learning with token selection for patient phenotyping from electronic health records (Sci Rep. 2020;10(1):1432) describes the development of DL models to construct a computable phenotype directly from the medical notes.
A large body of research has focused on extracting information from clinical notes in electronic health records. The approach can also be applied with some adjustment to other sets of unstructured data, including spontaneous reporting systems, as reflected in Identifying risks areas related to medication administrations - text mining analysis using free-text descriptions of incident reports (BMC Health Serv Res. 2019;19(1):791), product information documentation such as presented in Machine learning-based identification and rule-based normalization of adverse drug reactions in drug labels (BMC Bioinformatics. 2019;20(Suppl. 21):707) or even literature screening for systematic reviews as explored in Technology-assisted title and abstract screening for systematic reviews: a retrospective evaluation of the Abstrackr machine learning tool (Syst Rev. 2018 Mar 12;7(1):45).
In the systematic review Use of unstructured text in prognostic clinical prediction models: a systematic review (J Am Med Inform Assoc. 2022 Apr 27;ocac058), data extraction from unstructured text was shown to be beneficial in most studies. However, data extraction from unstructured text data does not show perfect accuracy (or related metric) and may have wide variability with respect to model performance for the same data extraction task, as shown in ADE Eval: An Evaluation of Text Processing Systems for Adverse Event Extraction from Drug Labels for Pharmacovigilance (Drug Saf. 2021;44(1):83-94). Thus, the application of these techniques should consider the objective in terms of precision or recall. For instance, a model that identifies medical concepts in a spontaneous report of an adverse drug reaction from a patient and maps it to a medical vocabulary might preferably focus on achieving a high recall, as false positives can be picked up in the manual review of the potential signal, whereas models with high precision and low recall may introduce irretrievable loss of information. In other words, ML models to extract data are likely to introduce some error and thus the error tolerance for the specific application needs to be considered.
16.5.2.2. Data insights
In pharmacoepidemiology, data insights extracted with ML models are typically one of three categories: confounding control, clinical prediction models and probabilistic phenotyping.
Propensity score methods are a predominant technique for confounding control (see Chapter 6.2.3.2). In practice, the propensity score is most often estimated using a logistic regression model, in which treatment status is regressed on observed baseline characteristics. In Evaluating large-scale propensity score performance through real-world and synthetic data experiments (Int J Epidemiol. 2018;47(6):2005-14) and A comparison of machine learning algorithms and covariate balance measures for propensity score matching and weighting (Biom J. 2019;61(4):1049-72) ML models were explored as alternatives to traditional logistic regression with a view to improve propensity score estimation. The theoretical advantages of using ML models include an automatisation procedure, by dispensing the need for investigator-defined covariate selection, and better modelling of non-linear effects and interactions. However, most studies in this field use synthetic or plasmode data and applications in real-world data need to be further explored.
The concept of rule-based, knowledge-based algorithms and risk-based stratification is not new to medicine and healthcare, the Framingham risk score being one of the most well-known. Trends in the conduct and reporting of clinical prediction model development and validation: a systematic review (J Am Med Inform Assoc. 2022;29(5):983-9) shows that there is a growing trend to develop data-driven clinical prediction models. However, the problem definition is often not clearly reported, and the final model is often not completely presented. This trend was exacerbated with the COVID-19 pandemic, where over 400 papers on clinical prediction models were published (see Prediction models for diagnosis and prognosis of covid-19: systematic review and critical appraisal, BMJ. 2020;369:m1328). The authors also suggest that prediction models are poorly reported, and at high risk of bias such that their reported predictive performance is probably optimistic, which was confirmed for several models in Clinical prediction models for mortality in patients with covid-19: external validation and individual participant data meta-analysis (BMJ. 2022;378:e069881). This is common, as has been reported in External validation of new risk prediction models is infrequent and reveals worse prognostic discrimination (Journal of Clinical Epidemiology. 2015;68(1):25–34.). While guidelines for reporting that are specific for AI prediction models are still under development (Protocol for development of a reporting guideline (TRIPOD-AI) and risk of bias tool (PROBAST-AI) for diagnostic and prognostic prediction model studies based on artificial intelligence, BMJ Open 2021;11:e048008), the Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD) statement can be used (BMJ 2015;350:g7594). Further, PROBAST: A Tool to Assess the Risk of Bias and Applicability of Prediction Model Studies (Ann Intern Med. 2019;170:51-58) supports the evaluation of prediction models. A review of checklists for reporting AI use is reported in Time to start using checklists for reporting artificial intelligence in health care and biomedical research: a rapid review of available tools (2022 IEEE 26th International Conference on Intelligent Engineering Systems (INES), IEEE 2022. p. 000015–20). A checklist for assessing bias in a ML algorithm is provided in A clinician's guide to understanding and critically appraising machine learning studies: a checklist for Ruling Out Bias Using Standard Tools in Machine Learning (ROBUST-ML) (European Heart Journal - Digital Health. 2022;3(2):125–40).
Clinical prediction models have also been applied for safety signal detection with some degree of success as exemplified in A supervised adverse drug reaction signalling framework imitating Bradford Hill's causality considerations (J Biomed Inform. 2015;56:356-68). For the evaluation of safety and utility, the Reporting guideline for the early stage clinical evaluation of decision support systems driven by artificial intelligence: DECIDE-AI can be used (BMJ 2022;377:e070904).
Probabilistic phenotyping is another potential use of ML in pharmacoepidemiology. It refers to the development of a case definition using a set of labelled examples to train a model and the outputting of the probability of a phenotype as a continuous trait. It differs from ML-based computable phenotyping mentioned earlier, as probabilistic phenotyping takes a set of features and estimates a probability of a phenotype whereas for the computable phenotyping, the ML technique merely extracts information that identifies a relevant case.
Methods for diagnosis phenotyping are discussed in Methods for Clinical Evaluation of Artificial Intelligence Algorithms for Medical Diagnosis. (Radiology. 2023 Jan;306(1):20–31). Validation of phenotyping of outcomes in pharmacoepidemiology, but not specifically AI related, is discussed in Core concepts in pharmacoepidemiology: Validation of health outcomes of interest within real-world healthcare databases (Pharmacoepidemiology and Drug Safety. 2023;32(1):1–8).
Identifying who has long COVID in the USA: a machine learning approach using N3C data (Lancet Digit Health. 2022;S2589-7500(22)00048-6) describes the development of a probabilistic phenotype of patients with long COVID using ML models and showed a high accuracy. Probabilistic phenotyping can be applied in wider contexts. In An Application of Machine Learning in Pharmacovigilance: Estimating Likely Patient Genotype From Phenotypical Manifestations of Fluoropyrimidine Toxicity (Clin Pharmacol Ther. 2020; 107(4): 944–7), a ML model using clincal manifestations of adverse drug reactions is used to estimate the probability of having a specific genotype, known to be correlated with severe but varied outcomes.
As development of probabilistic phenotypes is likely to increase, tools to assess the performance characteristics such as PheValuator: Development and evaluation of a phenotype algorithm evaluator (J Biomed Inform. 2019;97:103258) become more relevant.
Another possible category of use is hypothesis generation in causal inference, but this requires further research. For instance, in Identifying Drug-Drug Interactions by Data Mining: A Pilot Study of Warfarin-Associated Drug Interactions (Circ Cardiovasc Qual Outcomes. 2016;9(6):621-628) known warfarin–drug interactions and unknown possible interactions were identified using random forests.
16.5.3. Explainable AI
As AI decisions, predictions, extractions and other output can be incorrect, and sometimes especially so for a subgroup of people, it can cause risks and ethical concerns that must be investigated. As deep learning models are not directly interpretable, methods to explain their decisions have been developed. However, these provide only an approximation that might not resemble the underlying model and the performance is rarely tested.
In The false hope of current approaches to explainable artificial intelligence in health care (Lancet Digit Health. 2021;3(11):e745-e750), the authors show that incorrect explanations from current explainability methods can cause problems for decision making for individual patients, and they explain that these explainable AI methods are unlikely to achieve their asserted goals for patient-level decision support.
In Artificial intelligence in pharmacovigilance: A regulatory perspective on explainability (Pharmacoepidemiol Drug Saf. 2022;31(12):1308-1310) the authors argue that although by default pharmacovigilance models should require explainability, model performance may outweigh explainability in processes with high error-tolerance where, for instance, a human-in-the-loop is required, and the need for explainability should follow a risk-based approach.
16.6. Real-world evidence and pharmacoepidemiology
16.6.1. Introduction
The pharmacoepidemiology community has a long tradition of producing, evaluating, and interpreting observational data to provide evidence on the use, safety and effectiveness of medicines. The increasing ability to electronically capture and store data from routine healthcare systems and transform it into additional knowledge has opened up new opportunities for investigators to conduct studies. The terms real-world data (RWD) and real-world evidence (RWE) have been increasingly used since the early 2000’s to denote evidence generated from observational data collected during routine patient-level healthcare interactions. In medicines evaluation, evidence relying on RWD is now frequently submitted across the lifecycle of a product to complement and contextualise clinical trial knowledge with information from the routine healthcare setting, but the place of RWD in regulatory decision-making is still a subject of debate (see for example Replacing RCTs with real world data for regulatory decision making: a self-fulfilling prophecy? BMJ. 2023:380:e073100). Contribution of Real-World Evidence in European Medicines Agency’s Regulatory Decision Making (Clin Pharmacol Ther. 2023;113(1):136-51) reports that RWD/RWE was considered not supportive or was not further addressed in the regulatory evaluation report for 15 of 26 applications submitted to EMA in 2018-2019, where RWD/RWE was included to support efficacy pre-authorisation. Many issues discussed in the evaluation reports with respect to RWE were weaknesses related to methodological aspects, highlighting the need for adequate pharmacoepidemiological and statistical expertise in the generation of RWE.
There is currently no internationally agreed definition of RWD and RWE. Real World Evidence – Where Are We Now? (N Engl J Med. 2022;386(18):1680-2) emphasises that these terms are being used inconsistently and sometimes interchangeably across different health domains. Although evolving, a consistent terminology is yet to be established.
This chapter discusses ENCePP’s views on definitions of RWD and RWE, their role in medicines approval and evaluation, their relation to evidence generated by clinical trials, and why pharmacoepidemiological methods remain essential for the generation and assessment of RWD and RWE.
16.6.2. Definitions
The FDA’s Real-World Evidence website defines RWD as “the data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources” and RWE as “clinical evidence regarding the usage and potential benefits or risks of a medical product derived from analysis of RWD”. These definitions are widely used and have been adopted by other regulatory agencies. There is however a debate about the appropriateness of including both the nature of the data and the way it is collected (“routinely”) in the definition of RWD. RWD is commonly understood as observational data from various origins (e.g., electronic healthcare records, claims data, registries) but Marketing Authorization Applications Made to the European Medicines Agency in 2018-2019: What was the Contribution of Real-World Evidence? (Clin Pharmacol Ther. 2021;111(1):90-7) illustrates the difficulty of applying definitions of RWD in authorisation applications, notably when RWD is included as an element of clinical trials. Real-World Trends in the Evaluation of Medical Products (Am J Epidemiol. 2023;192(1):1-5) states that there is room for interpretation as to the data considered as RWD, for example data collected outside of health care settings for research purposes such as those collected through patient self-report outside of clinical encounters, or data collected through new technologies such as wearable biometric devices. This comment also applies to genetic data that are often collected outside the context of routine care or clinical trials but are generally considered as RWD. It is also noted that the data quality frameworks developed for RWD (see Chapter 13.2) examine how accurately the data represent the original information and how suitable they are but not how routinely they have been collected.
The view of ENCePP is that the specificity of RWD in comparison to any other observational data lies in the requirement for a true representation of the “real-world” patient characteristics (i.e., data with a high external validity) without influence of any specific study conditions. An assessment and validation of this real-world attribute, e.g., by external validation or benchmarking, is needed to provide assurance that it applies, or at least to evaluate and understand the deviation that may exist. A simpler definition of RWD could therefore only refer to patient data in contemporary clinical practice.
RWE is information derived from the analysis of RWD using sound epidemiological and statistical practices. The term RWE does not refer to specific methodologies and overlaps with pharmacoepidemiology, although it only partially overlaps with traditional classification of clinical research such as randomised vs. observational, prospective vs. retrospective or primary data collection vs. secondary use of data. The term RWE is nevertheless useful to state that the evidence originates from RWD, in the same way as the term experimental evidence is sometimes used to state that the evidence is based on experimental data.
16.6.3. Use of real-world evidence in medicines evaluation
There are many examples where RWD and RWE can be submitted to support medicines evaluation and regulatory decision-making. Three main objectives are identified in EMA’s DARWIN EU®: Multi-stakeholder information webinar (2022; slides 14-21):
-
to support the planning and validity of applicant studies, for example to inform the recruitment in pre- and post-authorisation studies, to examine the impact of planned inclusion/exclusion criteria, to measure the representativeness of the CT population (treatment and control arm) vs. the real-world target population and to evaluate whether the standard of care used in the control arm of a CT is comparable with the current real-word standard of care;
-
to understand the clinical context, for example to evaluate the incidence, prevalence and characteristics of diseases, to generate evidence on the actual clinical standards of care and compare them in different populations, and to characterise real-world drug use (incidence, prevalence, amount, duration, switching patterns);
-
to investigate associations and impact, for example to investigate the association between treatment exposure and either effectiveness or safety outcomes (including use of RWD as external control group), and to monitor the implementation and the effectiveness of risk minimisation measures.
Several studies have recently attempted to measure the frequency of use of RWD or RWE in marketing authorisation applications and the extent to which these data were actually utilised for decision-making, see, for example:
-
Use of real-world evidence in postmarketing medicines regulation in the European Union: a systematic assessment of European Medicines Agency referrals 2013-2017. BMJ Open 2019;9(10):e028133
-
Marketing Authorization Applications Made to the European Medicines Agency in 2018-2019: What was the Contribution of Real-World Evidence? Clin Pharmacol Ther. 2021;111(1):90-7
-
The Role of Real- World Evidence in FDA- Approved New Drug and Biologics License Applications. Clin Pharmacol Ther. 2022;111(1):133-44;
-
Use of Real-World Data and Evidence in Drug Development of Medicinal Products Centrally Authorized in Europe in 2018–2019. Clin Pharmacol Ther. 2022;111(1):310-20.
Due to variability in definitions, data sources, study designs and acceptability of RWD by regulatory decision-making bodies, very different estimates were found in these studies, with percentages of authorisation applications including RWE ranging from 39.9% to 100%.
How to enhance the suitability and acceptability of RWD/RWE to support authorisation applications is a matter of discussion and several publications have made proposals:
-
Contribution of Real-World Evidence in European Medicines Agency’s Regulatory Decision Making (Clin Pharmacol Ther. 2023;113(1):136-51) provides an in-depth analysis of the actual contribution of RWE in the decision-making on marketing authorisation approvals of applications submitted to EMA in 2018-2019, why such information was not considered supportive in some cases and how it contributed to the approval decision in other cases. It discusses suggestions to enable broader use of RWE in medicines development, including provision of data on mechanisms of action where RWE is used to extrapolate efficacy data from adults to children, previous experience with the medicinal product outside the EU application, description of the disease population and natural course of the disease, and early interactions (such as through scientific advice) between applicants and regulators to discuss the expected value of RWD to answer a specific research question, their limitations and how they could be minimised.
-
Harnessing Real-World Evidence to Advance Cancer Research (Curr. Oncol. 2023;30(2):1844-59) proposes a strategy with four steps: 1) to identify meaningful and well-defined clinical questions answerable with available RWD rather than scenarios for which RCTs are necessary and feasible; 2) to rely on high-quality RWD representative of the population of interest and contemporary clinical practice and with documented data completeness and provenance; 3) to use appropriate study designs accounting for data limitations, bias, confounding and sensitivity analyses; 4) to use clear, transparent and replicable study methodology to increase the confidence in the results.
-
Assessing and Interpreting Real-World Evidence Studies: Introductory Points for New Reviewers (Clin Pharmacol. 2022;111(1):145-9) details three aspects: the research question evaluated in the RWE study must align with the question of interest, with a recommendation to break it down according to the Population, Intervention, Comparator Outcome and Timing (PICOT) framework; the study design must use valid methods minimising selection bias, information bias and confounding, with a recommendation to use the target trial framework to help plan and design the RWE study; and the data must be suitable to address the research question, with elements of reliability (incl. plausibility and missingness) and relevance.
-
When Can We Rely on Real‐World Evidence to Evaluate New Medical Treatments? (Clin Pharmacol Ther. 2021;111(1):30-4) recommends that decisions regarding use of RWE in the evaluation of new treatments should depend on the specific research question, characteristics of the potential study settings and characteristics of the settings where study results would be applied, and take into account three dimensions in which RWE studies might differ from traditional clinical trials: use of RWD, delivery of real-world treatment and real-world treatment assignment.
-
Real-world evidence to support regulatory decision making: New or expanded medical product indications (Pharmacoepidemiol Drug Saf. 2021;30(6):685-93) reviews more specifically study designs used to generate RWE, including pragmatic trials, externally controlled trials and non-randomised healthcare database studies, among others.
-
Real-World Data for Regulatory Decision Making: Challenges and Possible Solutions for Europe (Clin Pharmacol Ther. 2019; 106(1):36-9) specifies four criteria for acceptability of RWE for regulatory purposes: it should be derived from data sources of demonstrated good quality, valid (with both internal and external validity), consistent (or heterogeneity should be explained) and adequate in terms of amount of information provided.
-
When and How Can Real World Data Analyses Substitute for Randomized Controlled Trials? (Clin Pharmacol. Ther. 2017;102(6):924-33) suggests that RWE is likely to be preferred over RCTs when studying a highly promising treatment for a disease with no other available treatments, where ethical considerations may preclude randomising patients to placebo, particularly if the disease is likely to result in severely compromised quality of life or mortality. In these cases, RWE could support product regulation by providing evidence on the safety and effectiveness of the therapy against the typical disease progression observed in the absence of treatment.
-
Reporting to Improve Reproducibility and Facilitate Validity Assessment for Healthcare Database Studies V1.0 (Pharmacoepidemiol Drug Saf. 2017;26(9):1018-32) highlights that substantial improvement in reproducibility, rigor and confidence in RWE generated from healthcare databases could be achieved with greater transparency about study parameters used to create analytic datasets from longitudinal healthcare databases and provides lists of specific parameters to be reported to increase reproducibility of studies.
Regulatory agencies have also published methodological recommendations to medicines developers on the submission of RWD/RWE within their applications to support their evaluation and acceptability:
-
Among other guidance available on the FDA’s Real-World Evidence website, a draft FDA guidance for industry provides Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products(2021), the draft guidance Real-World Data: Assessing Electronic Health Records and Medical Claims Data To Support Regulatory Decision-Making for Drug and Biological Products (2021) provides recommendations in three domains: data sources (relevance of data source and data capture), study design elements (time period, study population, exposure, outcome, covariates) and data quality, and the draft guidance Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products (2023) provides recommendations to sponsors and investigators considering the use of externally controlled clinical trials to provide evidence of the safety and effectiveness of a drug product.
-
The MHRA guidance on the use of real-world data in clinical studies to support regulatory decisions (2021) emphasises the importance of the quality of the data source, including its accuracy, validity, variability, reliability and provenance, with areas of consideration prior to submitting the study protocol. The MHRA guideline on randomised controlled trials using real-world data to support regulatory decisions (2021) provides points to consider when planning a prospective randomised trial using RWD sources with the intention of using the trial to support a regulatory decision, together with examples of scenarios, endpoints and designs.
-
Health Canada’s Elements of Real-World Data/Evidence Quality throughout the Prescription Drug Product Life Cycle (2019) provides overarching principles to guide the generation of RWE and an overview of some of the elements that should be addressed in protocol development and documentation of data quality within submissions containing RWE.
-
The Guidance for Reporting Real-World Evidence (2023) lays the foundation for the use of RWE in regulatory approval and Health Technology Assessment (HTA) in Canada; it focusses on guiding the evaluation of a study, providing core reporting standards and prioritising transparency in reporting while accounting for practical challenges related to RWD and RWE.
-
The EMA’s Guideline on registry-based studies (2021) provides recommendations on key methodological aspects that are specific to the use of patient registries by marketing authorisation applicants and holders planning to conduct registry-based studies for regulatory purposes.
16.6.4. Real-world evidence vs. clinical trials
The value of RWE to provide valid evidence on medicinal products as compared to clinical trials is a frequent subject of debate in the context of regulatory assessments, especially for medicines effectiveness where a departure from traditional clinical trials has been called on to speed-up their pace, reduce their cost and increase their generalisability. While RCTs are the gold standard for demonstrating the efficacy of medicinal products, they rarely measure the benefits and risks of an intervention when used in contemporary clinical practice and the current thinking is moving away from the long-held position that RWE is always inferior due to the likelihood of bias. Randomized Controlled Trials Versus Real World Evidence: Neither Magic Nor Myth (Clin Pharmacol Ther. 2021;109(5):1212–8) illustrates that the question is not about RCTs vs. RWE but about RCTs and RWE. In other words, use of observational evidence should generally not be considered to replace RCT information, except in specific circumstances, but both are complementary, as RWE may provide additional data, such as on longer follow-up of interventions and on treatment effects in populations not included in RCTs. Real World Evidence – Where Are We Now? (N Engl J Med. 2022;386(18):1680-2) suggests that randomised, non-randomised interventional and non-randomised non-interventional studies may rely on RWD for different objectives and therefore generate RWE, as illustrated by the following diagram:
{kind=link}
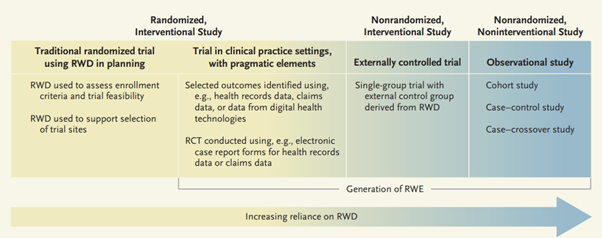
Reliance on RWD in Representative Types of Study Design. RCT denotes randomized, controlled trial; RWD real-world data; and RWE real-world evidence. Source: Concato J, Corrigan-Curay JD. Real World Evidence – Where Are We Now? (N Engl J Med. 2022;386(18):1680-2).
Statistical Considerations When Using RWD and RWE in Clinical Studies for Regulatory Purposes: A Landscape Assessment (Statistics in Biopharmaceutical Research 2023;15:1,3-13) discusses examples of when RWD can be incorporated into the design of various study types, including RCTs and purely observational studies, and reviews biostatistical challenges and methods for the use of RWE for medicinal product development.
A current domain of research is the assessment of whether non-interventional RWE studies can provide the same results as RCTs performed for the same research question. Emulation of Randomized Clinical Trials With Nonrandomized Database Analyses: Results of 32 Clinical Trials (JAMA 2023;329(16):1376-85) concludes that RWE studies can reach similar conclusions as RCTs when design and measurements can be closely emulated, but this may be difficult to achieve. Concordance in results varied depending on the agreement metric. Emulation differences, chance, and residual confounding can contribute to divergence in results and are difficult to disentangle.
16.6.5. Real-world evidence and pharmacoepidemiology
All the elements cited above to generate valid and reliable RWE using RWD are related to fundamental principles of pharmacoepidemiology. The widespread use of the concept of RWD/RWE has stimulated the use, accessibility and quality control of data sources as well as methodological developments to prevent and control bias and confounding, for example confounding by indication. Pharmacoepidemiologists should therefore take a leadership role and embrace this concept as a domain of research supporting regulatory decisions on medicinal products and public health in general. The following list includes areas of pharmacoepidemiological expertise that ENCePP considers important to develop and disseminate:
- Knowledge about RW data source metadata and its characteristics
- Understanding of different data types (e.g., primary care, specialist care, hospital care, disease registries, claims data, longitudinal drug prescription, dispensing or other drug utilisation data).
- Understanding of the context in which the data are collected, which should include – but not be limited to – local diagnostic criteria, local prescribing practices, local prescribing formularies, local coding practices, reimbursement policies, etc.
- Understanding of real-world data sources, including:
- common coding terminologies for drug exposure and clinical events,
- common data models,
- assessment of data quality (incl. data quality metrics, data quality frameworks, misclassification and missingness, benchmarking),
- their limitations and the statistical approaches to address them
- Knowledge about appropriate methods to establish meaningful RW evidence
- Expertise in epidemiological study designs, including traditional designs as well as case-only and case-population designs; studies with primary data collection vs. secondary use of data; prevalent-user vs. incident-user designs, positive and negative control exposures and outcomes; use of active exposure vs. non-exposure comparator groups.
- Knowledge of mechanisms of bias in observational studies (information bias, selection bias, confounding) and methods to address them at the design and analytical stages (incl. restriction, matching, stratification, modelling, use of propensity score methods, multiple imputation); methods to address unmeasured confounding and time-dependent confounding.
- Knowledge in handling effect modification, interaction and heterogeneity in observational studies.
- Expertise in assessing and validating different exposures, outcomes and covariates in observational studies.
- Knowledge in causal inference methods (incl. missing data handling, target trial emulation and interplay with ICH E9 (R1)).
- Knowledge in evidence synthesis, meta-analysis and data pooling.
- Experience in assessing a statistical analysis plan for an RWE study.